Toevallen/Epilepsie
Toevallen/Epilepsie admin 22 april, 2010 - 10:58Epilepsie. Algemeen
Epilepsie. Algemeen fajenn 6 mei, 2010 - 09:37Inleiding
InleidingEpilepsie is de naam voor een groep van ziekelijke aandoeningen die als gemeenschappelijk kenmerk het plotseling optreden van voorbijgaande abnormale verschijnselen hebben. Een niet-medische term voor deze aandoeningen is “vallende ziekte”.
De aanvallen zijn het gevolg van het gelijktijdig overmatig ontladen van neuronen in de hersenen. De aanvalsverschijnselen verschillen naar gelang de populatie van overmatig ontladende neuronen; ze kunnen motorisch, zintuiglijk, vegetatief, en/of psychiatrisch zijn en gepaard gaan met daling van bewustzijn. Zenuwcellen in de hersenschors kunnen gegeneraliseerd overmatig ontladen; dan ontstaan gegeneraliseerde aanvallen. Ze kunnen ook focaal overmatig ontladen en partiële aanvallen veroorzaken. Eén enkele aanval kan onder bijzondere omstandigheden bij iedereen voorkomen en is niet voldoende voor de diagnose epilepsie. Epilepsie kan leiden tot stoornissen van cognitie en gedrag in aanvalsvrije perioden. De behandeling van epilepsie kan neveneffecten hebben voor cognitie en gedrag.
Het aantal vormen van epilepsie is groot; de classificatie van de verschillende epilepsieën stuit op grote problemen. Overeenstemming is nog steeds niet bereikt. Deze tekst geeft een overzicht van de vele vormen van epilepsie en noemt de gevolgen voor cognitie en gedrag. Aan de effecten van medicamenteuze en chirurgische behandeling op cognitie en gedrag wordt aandacht gegeven.
We zullen eerst informatie geven over andere sites. Daarna komen aan de orde de oorzaken, de aanvalsverschijnselen, de epilepsieën die op geregelde wijze binnen een bepaalde context optreden (epilepsiesyndromen), het stellen van de diagnose, de behandelingen, psychosociale problemen, kwaliteit van leven en begeleiding
Op veel voorkomende of bijzondere vormen van epilepsie en op problemen van kinderen met deze epilepsieën wordt afzonderlijk ingegaan, zie:
Ziektebeschrijving Absences,
Ziektebeschrijving Rolandische epilepsie,
Ziektebeschrijving Temporale epilepsie,
Ziektebeschrijving Frontale epilepsie,
Ziektebeschrijving Syndroom van Landau Kleffner,
Ziektebeschrijving Kwaadaardige epilepsiesyndromen.
Andere teksten op internet
Andere teksten op internet- www.kinderneurologie.eu biedt uitvoerige en gedetailleerde Nederlandstalige informatie over een groot aantal vormen van epilepsie.
- www.ilae-epilepsy.org/ctf/ is de Engelstalige website van het internationale verbond tegen epilepsie. De tekst biedt deskundige medische informatie over vele vormen van epilepsie.
- www.epilepsie.nl bevat informatie voor patiënten en familieleden die voor het leven met epilepsie van belang zijn. Aangeboden door het Nationaal Epilepsiefonds.
- www.epilepsie.net Via dit adres kunnen patiënten en familieleden van patiënten met elkaar in contact komen en informatie verkrijgen.
- www.lwoe.nl wijst de weg naar advies over onderwijs
Epidemiologie
EpidemiologieOp basis van registraties door huisartsen in Nederland werd geschat dat in het jaar 2000 het aantal kinderen met epilepsie in de leeftijdsperiode van nul tot 14 jaar, afhankelijk van leeftijd en geslacht varieerde van 110 tot 240 per 100.000 (zie Peters & Gijsen, 2004, www.nationaalkompas.nl ). Epilepsie wordt genoemd als de meest voorkomende neurologische aandoening bij kinderen.
Oorzaken
OorzakenEen groot aantal aandoeningen leidt (mede) tot epilepsie, zoals blijkt uit het hier volgende lijstje (gewijzigd naar Loddenkemper et al. 2005, zie Jennekens-Schinkel en Jennekens, 2008)
Tabel
- genetische afwijkingen
- ontwikkelingsstoornissen van de hersenen
- hippocampussclerose
- tumoren
- vasculare malformaties
- infecties/ontstekingen van het centrale zenuwstelsel
- hersenbeschadiging door hypoxie-ischaemie
- hersenbeschadiging door stofwisselingsstoornissen of vergiftigingen
- hersentrauma
- structuurafwijkingen van de hersenen door onbekende oorzaak
- onbekende oorzaak
- Voor de kinderleeftijd zijn vooral ontwikkelingsstoornissen (dysplasieën), genetische afwijkingen en hippocampussclerose van belang.
- Dysplasieen kunnen het gevolg zijn van; (1) stoornissen bij de aanmaak van nieuwe hersencellen; (2) bij de migratie van nieuwe hersencellen naar de plaats van functioneren; en (3) tijdens de organisatie fase (zie ENCYCL- Anatomie van de hersenen). Het aantal mogelijke dysplasieën is verbluffend groot (Barkovich et al. 2012). Ze ontstaan door een genetische afwijking of door vooralsnog onbekende oorzaak. Ze kunnen de hele hersenschors betreffen maar ook zo klein zijn dat ze zelfs met optimale MRI technieken niet met zekerheid uitgesloten kunnen worden.
- Genetische afwijkingen kunnen oorzaak zijn van hersenziekten die met epilepsie gepaard gaan (zie Tabel) en van epilepsiesyndromen.
- Hippocampussclerose. De oorzaak van hippocampussclerose is vaak duister; in sommige gevallen zou verband kunnen bestaan met eerder doorgemaakte, langdurige koortsstuipen (Jennekens-Schinkel & Jennekens, 2008). - Bij sommige hersenaandoeningen komen de aanvallen voor in het kader van een complex van verschijnselen, bijvoorbeeld alleen in een bepaalde levensfase, vooral 's nachts, gecombineerd met een spraakstoornis en speekselvloed. Men spreekt in die gevallen over “epilepsiesyndromen”. Van sommigen daarvan is een genetische basis aangetoond.
Aanvallen
AanvallenKenmerken van epileptische aanvallen
- Epileptische krampaanvallen of trekkingen kunnen tonisch zijn of klonisch. Een tonische aanval bestaat uit tenminste 3 seconden aanhoudende contractie(s) van spieren; klonische aanvallen verlopen ritmisch en volgen vaak op de tonische (dan spreekt men van tonisch-klonische aanval). De trekkingen kunnen zich gegeneraliseerd maar ook partieel voordoen. Myoklonieën - spiertrekkingen, soms verspreid voorkomend - zijn niet altijd epileptisch.
- Absences zijn kortdurende bewustzijnsdalingen die met minimale motorische verschijnselen gepaard gaan en die kenmerkende afwijkingen op het eeg tonen.
- Aura’s zijn het allereerste of enige verschijnsel van een aanval, alleen het kind zelf, niet de omgeving) neemt het waar.
- Wanneer de aanvallen snel achtereen terugkeren zonder duidelijke tekenen van beëindiging tussen de aanvallen, dan spreekt men van een status epilepticus.
- Het aanhoudend terugkeren van trekkingen in een deel van het lichaam, bijvoorbeeld in een arm, wordt epilepsia partialis continua genoemd.
- Epileptische spasmen zijn plotselinge houdingsveranderingen van de romp - buigen of strekken - meestal met buiging van het hoofd en strekken van de ledematen. Spasmen komen vooral voor in de eerste levensjaren.
Aanvallen van pasgeborenen: neonatale convulsies Verschillende soorten trekkingen kunnen zich bij pasgeborenen voordoen. Ze zijn niet allemaal epileptisch. Neonatale convulsies kunnen de aanwezigheid signaleren van hersenafwijkingen, zie Ziektebeschrijving Zuurstoftekort rond de geboorte, Ziektebeschrijving Beroerte rond de geboorte, en Ziektebeschrijving Bacteriële meningitis. Er is ook een autosomaal dominant overervende vorm van neonatale epilepsie. Men neemt doorgaans aan dat de aanvallen een ongunstig effect hebben op het ontwikkelende hersenweefsel. De beste behandeling staat nog niet vast (Glass et al., 2009). De gevolgen voor cognitie en gedrag hangen overwegend af van de aandoening die aan de aanvallen ten grondslag liggen. Focale aanvallen De aanvallen verschillen afhankelijk van de leeftijd van het kind en de plaats van het focus.
- Karakteristieken. Naar schatting is bijna de helft van alle epileptische aanvallen in de kindertijd focaal. De aanvalsverschijnselen verschillen naar gelang de groep van zenuwcellen die gelijktijdig ontlaadt. De verschijnselen hangen mede af van de leeftijd van het kind. Bij kinderen vanaf ongeveer 6 jaar zijn de aanvallen zoals bij volwassenen. Bij jongere kinderen (< 6 jaar) gaan focale aanvallen vaker dan bij oudere kinderen gepaard met motorische verschijnselen. De aanvallen beginnen vaak met een aura. Frontale aanvallen gaan vaak gepaard met bewegingen, temporale aanvallen met gedragsonderbreking en occipitale aanvallen met al of niet gekleurde lichtflitsen en andere abnormale visuele waarnemingen. Het overmatig ontladen kan zich verbreiden naar nabijgelegen zenuwcellen: de partiële aanval komt dan, secundair, tot generalisatie. Van de vele verschillende oorzaken voor focale aanvallen komen tumoren en aangeboren misvormingen het meest voor (Jennekens-Schinkel & Jennekens, 2008).
- Cognitie en gedrag bij focale aanvallen. Het intelligentiequotiënt (IQ) is bij kinderen met frontale aanvallen doorgaans licht verlaagd. In groepen kinderen met goed behandelbare temporale aanvallen is het IQ ook licht verlaagd. Bij temporale epilepsie kunnen selectieve stoornissen in enkele cognitieve functies voorkomen: bijvoorbeeld bemoeilijkt herkennen van gezichten bij temporale laesies in de rechterhemisfeer. Kinderen met moeilijk behandelbare, in het eerste levensjaar begonnen temporale epilepsie zijn vaak zwakzinnig (ENCYCL-Zwakzinnigheid) (Zie Ziektebeschrijving Temporale epilepsie).
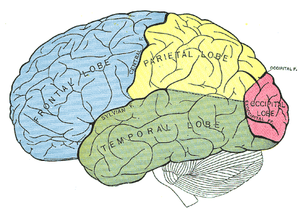
Figuur overgenomen via Google afbeeldingen van www.nl.wikipedia.org/
Figuur overgenomen via Google afbeeldingen van www.evodisku.multiply.com
Aanvallen tijdens koorts
Koortsstuipen zijn trekkingen in armen en benen die (1) ten hoogste 15 minuten aanhouden, (2) optreden bij koorts (> 38 graden) (3) bij een kind in de leeftijd tussen 3 maanden en 6 jaar dat (4) geen neurologische aandoening heeft. Er is verband tussen de ontwikkelingsgraad van de hersenen en het verschijnsel koortsstuipen. Koortsstuipen worden niet als epileptische aanvallen beschouwd. Wel is de kans op epilepsie later in het leven bij deze kinderen iets verhoogd. Behandeling van koortsstuipen heeft meer nadelen dan voordelen (Steering Committee, 2008).
Koortsstuipen zijn “complex” als ze (1) langer dan 15 minuten aanhouden, (2) partieel zijn, (3) binnen 24 uur en/of vaak terugkomen. Koortsstuipen na het 6de jaar worden aangeduid als FS+ (febrile seizures plus). Bij complexe koortsstuipen en FS+ is het risico op het ontstaan van epilepsie groter dan bij andere koortsstuipen. Koortsstuipen komen voor bij 2-5% van de kinderen (Steering Committee , 2008).
Koortsstuipen hebben op zichzelf geen gevolgen voor de cognitieve ontwikkeling. Bij koortsstuipen van jonge kinderen die lang aanhouden (> 30 minuten) moet rekening worden gehouden met tekorten in taken die beroep doen op herkenning van eerder waargenomen voorstellingen zoals afbeeldingen van gezichten. Dit is aangetoond tot een jaar na de koortsstuipen. De betekenis van deze bevinding voor het latere functioneren is nog onbekend. Onzeker is of het tekort het gevolg is van de koortsstuipen zelf dan wel van de aandoening die oorzaak is van de ongewoon lang aanhoudende stuipen (Martinos et al. 2012)
Epilepsie syndromen
Epilepsie syndromenHet aantal epilepsiesyndromen is groot. Men onderscheidt goedaardige en kwaadaardige syndromen. Er is ook een tussencategorie. Een syndroomdiagnose kan helpen bij het doen van een uitspraak over het toekomstig beloop. Het is lang niet altijd mogelijk de oorzaak van epilepsie of het epilepsiesyndroom vast te stellen
Goedaardige syndromen
Tenminste 9 verschillende syndromen zijn bekend (Jennekens-Schinkel & Jennekens, 2008).
(1-5) Twee syndromen beginnen neonataal, twee andere in het eerste levensjaar, en een vijfde syndroom begint in de eerste twee levensjaren. De aanvallen verdwijnen spontaan of reageren goed op anti-epileptica. Na verdwijnen van de aanvallen bestaat een verhoogde kans op epilepsie in latere kinderjaren. Drie van deze vijf epilepsiesyndromen zijn autosomaal dominant erfelijk.
(6) Syndroom van Panayiotopoulos. In de kinderjaren, vooral tussen 3 en 6 jaar. Aanvallen van bewustzijnsdaling en verwardheid, voorafgegaan door misselijkheid, braken, laten lopen van urine of ontlasting. Aanvallen komen sporadisch voor, vooral ’s nachts, en verdwijnen na één tot twee jaar. Een minderheid van de kinderen krijgt daarna voorbijgaand een andere vorm van epilepsie, meestal Rolandische epilepsie.
(7) Rolandische Epilepsie of Benigne Epilepsie met Centrotemporale Pieken bij Kinderen. Dit is de meest voorkomende vorm van epilepsie in de schooljaren. De aanvallen beperken zich tot een deel van het lichaam en komen vooral ’s nachts voor. Zie Ziektebeschrijving Rolandische epilepsie.
(8) Absence epilepsie beginnend in de kinderjaren of juveniel. Zie Ziektebeschrijving Absence epilepsie.
(9) Juveniele myoklonus epilepsie. De aanvallen beginnen na het 10de jaar. Het zijn vooral myoklonieën maar absences en tonisch-klonische aanvallen komen ook voor. Vaak bestaat epilepsie bij familieleden. De jongeren kunnen jarenlang aanvallen houden.
Cognitie en gedrag. De goedaardige syndromen veroorzaken geen of lichte cognitieve problemen en zijn vrijwel altijd verenigbaar met normaal basisonderwijs. De kinderen kunnen in het dagelijkse leven normaal functioneren maar het vergt wat meer “herseninspanning” dan bij gezonde leeftijdgenoten (Myatchin et al., 2009).
Syndromen met variabel beloop
Kwaadaardige syndromen
Myoklonische encefalopathie (1), en de syndromen van Ohtahara (2), West(3), Lennox-Gastaut (4), Doose (5) en Dravet(6) hebben veelvormige epileptische aanvallen en karakteristieke afwijkingen in het electro-encefalogram. De aanvallen beginnen in de eerste levensmaanden of in de vroege kinderjaren. Bij veel kinderen met deze syndromen is een ernstige hersenaandoening aantoonbaar, soms is als oorzaak een gen-afwijking het meest aannemelijk. Zie Ziektebeschrijving Kwaadaardige epilepsiesyndromen.
Cognitie en gedrag De kinderen blijven achter in ontwikkeling of gaan in ontwikkelingsniveau achteruit.
Behandeling
BehandelingAnti-epileptica
Met chemische middelen kunnen aanvallen worden beheerst, niet genezen. Over de indicatie tot behandeling, het tijdstip van aanvang van behandeling, de keuze uit een grote reeks voorhanden anti-epileptica en de dosering, bijwerkingen en duur van de behandeling met anti-epileptica vindt voortdurend onderzoek en overleg plaats. Negen tot vierentwintig procent van de kinderen bereikt geen aanvalsvrijheid (Raspall-Chaure et al., 2008). De International League Against Epilepsy oordeelde in 2013 over het onderzoek van anti-epileptica dat “het gebrek aan goed opgezette ... onderzoeken ... bij kinderen is alarmerend … (Glauser et al., 2013).Tot de anti-epileptica die in eerste instantie worden voorgeschreven behoren lamotrigine en carbamazepine (zie Basiscursus Epilepsie, 2009; Aylward, 2008).
Cognitie en gedrag
- De klassieke anti-epileptica hebben ongunstige cognitieve en gedragsmatige neveneffecten. Dat geldt bijvoorbeeld voor het effect van phenobarbital op het IQ.
- Recent ontwikkelde anti-epileptica hebben deze nevenwerkingen in veel mindere mate maar kunnen niet over één kam geschoren worden. Bij volwassenen verbeteren de scores van neuropsychologisch testonderzoek zeer aanmerkelijk na staken van anti-epileptica (met name carbamazepine en valproaat) wegens verkregen aanvalsvrijheid, zo bleek uit grote Noorse studies (Hessen et al ., 2009).
- Lamotrigine wordt geacht weinig effecten op de cognitie te hebben.
- Levetiracetam is bij volwassenen klaarblijkelijk niet helemaal zonder nadelige cognitieve effecten; zowel lamotrigine als levetiracetam hebben op het cognitieve functioneren van volwassenen een minder belemmerende werking dan carbamazepine (Meador 2008).
- Verschillende studies wijzen op meer nadelige cognitieve effecten van topiramaat dan van levetiracetam (Meador 2008). Topimaraat heeft een nadelige invloed op woordvinding.
De ernst van de effecten hangt af van de dosering. Onder de andere punten die mogelijk van invloed zijn op cognitie en gedrag, moeten worden gerekend de leeftijd waarop met behandeling wordt begonnen, de duur van het gebruik van anti-epileptica, en het gelijktijdige gebruik van verschillende anti-epileptica (polytherapie). De werking van de anti-epileptica op cognitie en gedrag moet uiteraard worden onderscheiden van a) de gevolgen die de aandoening heeft die de epilepsie veroorzaakt en b) de mogelijk nadelige gevolgen van de epileptische aanvallen (Raspall-Chaure et al., 2008).
Ketogeen dieet
Een hoog ketongehalte van het bloed kan gunstig effect hebben op de epilepsie van kinderen, ook als anti-epileptica niet effectief zijn. Deze overtuiging bestaat al tientallen jaren en is in 2008 voor het eerst bevestigd in een gerandomiseerde, gecontroleerde, niet-blinde studie (Neal et al., 2008). Een hoge ketonspiegel van het bloed kan verkregen worden met een vetrijk dieet dat arm is aan zetmeel en koolhydraten. Een ketogeen dieet blijkt bij een minderheid van de kinderen het aantal aanvallen en de ernst van de aanvallen te verminderen. Smakelijk is het dieet niet en lang niet alle kinderen houden het vol. Hoe lang het effect aanhoudt na staken van het dieet is niet bekend.
Cognitie en gedrag
Over gevolgen van het ketogene dieet voor cognitie en gedrag zijn niet voldoende gegevens beschikbaar voor definitieve richtlijnen, maar veel ouders vinden hun kinderen alerter, vaak een reden temeer om het dieet vol te houden.
Nervus vagus stimulatie
Deze behandeling wordt toegepast als anti-epileptica en dieet falen en epilepsiechirurgie niet in aanmerking komt of onvoldoende succes heeft. Bij het ontstaan van de epileptische aanval vindt een proces plaats van synchronisatie van activiteit van neuronen. Met nervus vagus stimulatie kan enige mate van desynchronisatie worden bereikt. De stimulator wordt via een kleine ingreep onderhuids in de hals ingebracht. Het effect op de epilepsie is overwegend onderzocht door achteraf (retrospectief) na te gaan hoe het met de aanvallen is gegaan, zonder te vergelijken met een controlegroep. Ter illustratie: van 34 kinderen was na een jaar één kind aanvalsvrij, het aantal aanvallen verminderde met meer dan 50% bij 13 kinderen en met minder dan 50% bij 20 kinderen (Sherman et al., 2008).
Cognitie en gedrag
Effect van n. vagus stimulatie op cognitie en gedrag is niet overtuigend aangetoond (Boon et al., 2006)
Epilepsiechirurgie
Epilepsiechirurgie wordt overwogen als epilepsie ernstig is en de behandeling met anti-epileptica geen of zeer onvoldoende succes heeft.
- De ingreep zal verenigbaar moeten zijn met behoud van essentiële functies, waaronder taal en geheugen. Dit kan onder andere worden onderzocht door kortdurende uitschakeling van een helft van de grote hersenen door middel van toediening van amobarbital (een barbituraat) aan het grote toevoerende bloedvat van die hersenhelft (de arteria carotis). De neuropsycholoog heeft tot taak in kort tijdsbestek zich een indruk te vormen van taal en geheugen in deze conditie. De amobarbital procedure staat (naar de bedenker ervan) bekend als Wada test. Tegenwoordig wordt ook electrocorticografie (EcOG) toegepast.
- De ingreep (verwijdering of uitschakeling van epileptisch hersenweefsel) kan plaatselijk (focaal) zijn, bijvoorbeeld een deel van de temporale gebieden (slaapkwab) of van frontale gebieden (voorhoofdskwab); ze kan ook een hele hersenhelft omvatten (hemisferectomie). De hersendelen die uitgeschakeld of verwijderd worden zijn meestal structureel abnormaal, bijvoorbeeld door aanwezigheid van een goedaardige tumor of van dysplasie van een gebied in de hersenschors. Hemisferectomie wordt overwogen bij aandoeningen die een groot deel van een hersenhelft omvatten en de andere helft sparen, zoals bijvoorbeeld vaatafwijkingen bij het syndroom van Sturge-Weber en ontstekingsachtige afwijkingen bij het syndroom van Rasmussen.
- Uitschakeling van een deel van de hersenen is niet zonder nadelen, ook als het verwijderde deel grotendeels abnormaal is. Zo kan het gezichtsveld na verwijdering van een deel van de temporaalkwab aan één zijde ingeperkt zijn. Na hemisferectomie neemt de doorgaans al bestaande zwakte in een lichaamshelft toe, met name van de hand.
- Na focale epilepsiechirurgie wordt ongeveer 60 – 70% van de kinderen aanvalsvrij en na hemisferectomieën 45 – 80%. Bij dysplasieën zijn de resultaten minder goed omdat deze vaak niet plaatselijk mooi afgebakend zijn en vaak ook in de andere hersenhelft voorkomen. Vrijheid van aanvallen wordt ervaren als een bevrijding en vergroot de mogelijkheden tot sociale participatie (van Empelen et al., 2005).
Cognitie en gedrag
- Van de kinderen die voor epilepsiechirurgie in aanmerking komen zijn de meeste zwakzinnig. Na operatie gaat de ontwikkeling niet verder achteruit; voor zover de ontwikkeling voor uitgaat is dat meestal minder dan normaal, onvoldoende om toename van achterstand te voorkomen (dat wil zeggen, niet anders dan in andere gevallen van zwakzinnigheid). Aanmerkelijke postoperatieve cognitieve vooruitgang komt bij hoge uitzondering voor (Spencer & Huh, 2008).
- Voorafgaande aan operaties van de temporale kwab vanwege temporale epilepsie hebben kinderen in de regel globale taalachterstand, dat wil zeggen zowel in spreken als in luisteren en zowel in de woordenschat als in de grammatica (ENCYCL-Taalverwerving). Na de operatie verloopt de taalontwikkeling bij een grote minderheid van de kinderen wat sneller dan normaal zodat de taalachterstand vermindert, bij velen is de taalontwikkeling echter langzamer dan normaal, zodat de achterstand toeneemt. Dat geldt in het bijzonder bij linkszijdige operaties wanneer de taak van de linkerhersenhelft bij het verwerken van taal niet is overgenomen door de rechterhersenhelft (Spencer & Huh, 2008; de Koning et al., 2009). Zie ook Ziektebeschrijving Temporale epilepsie.
Psychosociaal functioneren
Psychosociaal functionerenGedrag
Gegevens over gedragsproblemen zijn zeer overwegend verkregen met eenzelfde vragenlijst, de Child Behavior Checklist (CBCL). Kinderen met epilepsie hebben meer gedragsproblemen dan niet zieke kinderen en iets meer dan kinderen met andere chronische ziekten. De gedragsproblematiek van kinderen met kwaadaardige vormen van epilepsie verschilt sterk van die bij kinderen met de veel meer voorkomende overwegend goedaardige epilepsieën. Tot de veel voorkomende gedragsproblemen behoren onvoldoende kunnen opbrengen van aandacht, neiging tot teruggetrokkenheid, piekeren, verlegenheid en het niet tot afsluiting brengen van taken of handelingen. Kinderen met ernstiger vormen van epilepsie tonen verhoudingsgewijze meer agressief gedrag (Asato et al., 2009; Rodenburg et al., 2005;).
Schoolloopbaan
Een kind heeft een leerprobleem als de schoolprestaties achterblijven bij de verwachtingen op grond van de intelligentie van het kind. In het reguliere basisonderwijs komen doublures van een schooljaar en extra onderwijshulp veel meer voor bij kinderen met in de schoolleeftijd gediagnosticeerde epilepsie dan bij andere kinderen. De leerproblemen dateren al van de periode voordat de epilepsie is gediagnosticeerd en behandeling met anti-epileptica is begonnen (Hermann et al., 2008; Oostrom et al., 2005)).
Kwaliteit van leven
Kwaliteit van levenIn groepsdiscussies van schoolkinderen (leeftijd tussen 6 en 12 jaar) met epilepsie blijkt vooral bezorgdheid over hun acceptatie door leeftijdgenoten en voor de restricties die epilepsie meebrengt, bijvoorbeeld geen toestemming krijgen tot logeren bij leeftijdgenootjes, niet alleen mogen fietsen of sporten. Kinderen zijn bang voor nieuwe aanvallen en voor de risico’s die erdoor ontstaan. Ze hebben soms weerstand tegen de anti-epileptische medicatie.
Voor adolescenten zijn de wegens epilepsie opgelegde restricties in het bereiken van de zelfstandigheid die bij hun leeftijdsfase past vaak struikelblokken. Adolescenten zijn bezorgd over de gevolgen van de epilepsie voor hun toekomst (Moffat el al., 2009).
Begeleiding
BegeleidingBehandelaars en begeleiders van kinderen met epilepsie hebben niet alleen aandacht voor de aanvallen en de gevolgen daarvan maar ook voor andere manifestaties van de hersenaandoening die ten grondslag ligt aan de aanvallen en op de reacties van mensen in de omgeving van het kind met aanvallen.
- Een grote aanval met tonische en klonische trekkingen (‘grand mal’) kan bij opvoeders en omstanders schrik en vrees voor doodgaan van het kind doen ontstaan. Kleine aanvallen met kortdurende bewustzijnsdaling en minimale motorische verschijnselen (absences, ‘petit mal’) kunnen geïnterpreteerd worden als epileptische afwijkingen of ook wel als stoornissen in aandacht en concentratie. Nachtelijke epilepsie kan leiden tot vrees voor inslapen, tot slaapgebrek en tot nadelige gevolgen voor gedrag overdag. Behandelaars en begeleiders met kennis van de heterogeniteit van epileptische aanvallen en van de reacties daarop, kunnen over dit alles informatie geven en adviseren. Ze kunnen gedragsbeperkingen aanvaardbaar maken en vrees wegnemen.
- Epilepsie is geen teken van minderwaardigheid van het zenuwstelsel en nog minder van een persoon of familie, het is geen uiting van zondigheid, ze zou kinderen niet moeten stigmatiseren. Epilepsie is een ziekteverschijnsel, net als andere ziekteverschijnselen. Informatie aan ouders, aan leerkrachten, en aan kinderen in de klas helpt de omgeving om verstandig en meelevend om te gaan met kinderen die epilepsie hebben. Het Nationaal Epilepsie Fonds verzorgt samen met de patiëntenorganisatie Epilepsie Vereniging Nederland op verzoek voorlichtingsbijeenkomsten op scholen. Het Nationaal Epilepsie Fonds heeft een Infolijn waar ouders informatie kunnen vragen. Telefoon nummer 0900. 8212411. Zie ook de adressen bij Andere teksten op internet.
- Er is een landelijk dekkend netwerk van Steunpunten voor Onderwijs en Epilepsie met ambulante begeleiders die verbonden zijn aan de Centra De Waterlelie (Cruquius in Heemstede en in Zwolle) en De Berkenschutse in Heeze. Zie www.lwoe.nl
- In geval van klachten of onzekerheid over cognitie en gedrag kan neuropsychologisch onderzoek worden verricht, om (a) eventuele cognitieve beperkingen vast te stellen, (b) te adviseren over opvoeding, onderwijs en beroepskeuze, (c) afhankelijk van het verloop van de epilepsie bij herhaling van onderzoek uitgangsgegevens ter beschikking te hebben en eventuele bijwerkingen van AED op te sporen.
Literatuur
LiteratuurAsato MR, Manjunath B, Sheth RD, Phelps SJ, Wheless JW, Hovinga CA, Pina-Garza JE, Haskins LS, Zingaro WM (2009) Adolescent and caregiver experiences with epilepsy. Journal of Child Neurology 24: 562-573
Aylward RL (2008) Epilepsy: a review of reports, guidelines, recommendations and models for the provision of care for patients with epilepsy. Clinical Medicine 8: 433-438
Barkovich AJ, Guerrini R, Kuzniecky RI, Jackson GD, Dobijns WB (2012) A developmental and genetic classification of malformations of cortical development: update 2012. Brain 135: 1348 - 1369
Basiscursus Epilepsie. Stichting Epilepsie Onderwijs Nederland, SepiON, 2009
Boon P, Moors I, De Herdt V, Vonck K (2006) Vagus nerve stimulation and cognition. Seizure 15: 259-263
Empelen R van, Jennekens-Schinkel A, Rijen PC van, Helders PJM, Nieuwenhuizen O van (2005) Health-related quality of life and self-perceived competence of children before and up to two years after epilepsy surgery. Epilepsia 46: 258-271
Glass HC, Wirrell E (2009) Controversies in neonatal seizure management. Journal of Child Neurology 24: 591-599
Glauser T, Ben-Menachem E, Bourgeois B, Cnaan A, Guerreiro C, Kalviainen R et al. (2013) Updated ILAE evidence: review of anti epileptic drug efficacy and effectiveness as initial monotherapy for epileptic seizures and syndromes. Epilepsia 54: 551 - 563
Hessen E, Lossius MI, Gjerstad L (2009) Antiepileptic monotherapy significantly impairs normative scores on common tests of executive functions. Acta Neurologica Scandinavica 119: 194-198
Hermann B, Seidenberg M, Jones J (2008) The neurobehavioral comorbidities of epilepsy: can a natural history be developed. Lancet Neurology 7: 151-159
Jennekens-Schinkel A & Jennekens FGI (2008) Neuropsychologie van neurologische aandoeningen in de kindertijd. Amsterdam, Boom, Hoofdstuk 11, pp 450-484
Koning T de, Versnel H, Jennekens-Schinkel A, Schooneveld MMJ van, Dejonckere PH, Rijen PC van PC, Nieuwenhuizen O van: on behalf of the Dutch Collaborative Epilepsy Surgery Programme (DuCESP) (2009) Language development before and after temporal surgery in children with intractable epilepsy. Epilepsia: 50(11), 2408-2419
Martinos MM, Yoong M, Patil S, Chin RFM, Neville BG, Scott RC, de Haan M (2012) Recognition memory is impaired in children after prolonged febrile seizures. Brain 135: 3153 - 3164
Meador KJ (2008) Cognitive effects of levetiracetam versus topiramate. Epilepsy Currents 8: 64-65
Moffat C, Dorris L, Connor L, Espie CA (2009) The impact of childhood epilepsy on quality of life: A qualitative investigation using focus group methods to obtain children’s perspectives on living with epilepsy. Epilepsy & Behavior 14: 179-189
Myatchin I, Mennes M, Wouters H, Stiers P, Lagae L (2009) Working memory in children with epilepsy: an event-related potentials study. Epilepsy Research 86: 183-190
Neal EG, Chaffe H, Schwartz RH, Lawson MS, Edwards N, Fitzsimmons G, Whitney A, Cross JH (2009) A randomized trial of classical and medium-chain triglyceride ketogenic diets in the treatment of childhood epilepsy. Epilepsia 50: 1109-1117
Oostrom KJ, Teeseling H van, Smeets-Schouten A, Peters ACB, Jennekens-Schinkel A on behalf of the Dutch Study of Epilepsy in Childhood (DuSECh) (2005) Three to four years after diagnosis: cognition and behaviour in children with 'epilepsy only'. A prospective, controlled study. Brain 128: 1546-1555
Peters ACB, Gijsen (2004) Prevalentie, incidentie en sterfte naar geslacht en leeftijd. Nationaal Kompas, Epilepsie. www.nationaalkompas.nl
Raspall-Chaure M, Neville B, Scott RC (2008) The medical management of the epilepsies in children: conceptual and practical considerations. Lancet Neurology 7: 57-69
Rodenburg R, Stams GJ, Meijer AM, Aldenkamp AP, Deković M (2005) Psychopathology in children with epilepsy. Journal of Pediatric Psychology 30: 453-468
Sherman EMS, Connolly MB, Slick DJ, Eyrl KL, Steinbok P, Farrell K (2008) Quality of life and seizure outcome after vagus nerve stimulation in children with intractable epilepsy. Journal of Child Neurology 23: 991-998
Spencer S & Huh L (2008) Outcomes of epilepsy surgery in adults and children. Lancet Neurology 7: 525-537
Steering Committee on Quality Improvement and Management (2008) Febrile seizures: clinical practice guideline for the long-term management of the child with simple febrile seizures. Pediatrics 121: 1281-1286
Absence epilepsie
Absence epilepsie fajenn 6 mei, 2010 - 09:41Inleiding
Inleiding“Absences” zijn plotselinge kortdurende bewustzijnsdalingen die vaak met onopvallende andere verschijnselen (bijvoorbeeld even knipperen met de ogen) gepaard gaan en waarbij zich karakteristieke afwijkingen in het eeg voordoen. Typische absences komen voor bij “absence epilepsie in de kinderjaren” (childhood absence epilepsy, bekend als CAE) en bij “juveniele absence epilepsie” (juvenile absence epilepsy, of JAE). Epilepsie met absences kan na enige tijd overgaan in epilepsie met vooral spiertrekkingen (myoklonieën): men spreekt dan van juveniele myoklonische epilepsie (Jennekens-Schinkel & Jennekens, 2008).
Wij vatten eerst de lichamelijke verschijnselen van CAE en JAE samen en beschrijven daarna de afwijkingen in cognitie en gedrag bij CAE. Eenzelfde beschrijving is wegens gebrek aan voldoende gegevens niet mogelijk voor JAE.
Andere teksten op internet
Andere teksten op internet- www.kinderneurologie.eu is een aan te raden Nederlandstalige tekst bestemd voor (para)medici en anderen.
- www.ilae-epilepsy.org/ctf . Op de website van het internationale verbond tegen epilepsie bieden vooraanstaande deskundigen medische informatie over absence epilepsie.
- Zie voor patiënt gerichte informatie Ziektebeschrijving Epilepsie, andere teksten op internet,.
Epidemiologie
Epidemiologie“Absence epilepsie in de kinderjaren” vormt 6 tot 8% van alle epilepsieën bij kinderen van 15 jaar en jonger. Bij kinderen jonger dan 6 jaar bedraagt het percentage ongeveer 5 en bij kinderen van 6-10 jaar ongeveer 18 (Dura-Travé et al., 2008; Larsson et al., 2006). Absence epilepsie komt meer voor bij meisjes (60%) dan bij jongens. Van de epileptische kinderen van 10 – 15 jaar heeft ongeveer 10% juveniele absence epilepsie.
Oorzaken
OorzakenEen genetische factor als oorzaak van absence epilepsie is om verschillende redenen aannemelijk. Ten eerste heeft een grote minderheid van de kinderen met absence epilepsie ook familieleden met epilepsie. Ten tweede komt absence epilepsie veel meer voor bij ieder lid van eeneiige, genetisch identieke, tweelingen dan bij ieder lid van twee-eiige tweelingen (de zogeheten concordantie is groter bij een- dan bij twee-eiige tweelingen). DNA onderzoek wijst bij absence epilepsie op variaties in DNA sequenties in tenminste twee genen. Men veronderstelt dat dit de kans op absence epilepsie verhoogt (Chioza et al., 2009; Panayiotopoulos, 2008).
Lichamelijke verschijnselen
Lichamelijke verschijnselenIn de kinderjaren
De aanvallen duren omstreeks 8 seconden. Ze zijn vooral gekenmerkt door kortdurende bewustzijnsdaling, dat wil zeggen onderbreking van “besef” en onderbreking van de activiteit van het kind. Het kind reageert niet op aanspreken; na afloop van de absence hervat het zijn activiteit waar het gebleven was en zonder te weten wat zich heeft afgespeeld. Tijdens de absence kunnen zich onopvallende motorische of vegetatieve verschijnselen voordoen: onder andere myoklonieën in oogleden of wenkbrauwen of rond de mond, pupilverwijding en versnelde hartactie. De meeste kinderen hebben dagelijks absences, sommigen zelfs honderden per dag. Tijdens de aanvallen worden in het eeg over de gehele hersenoppervlakte gelijktijdig links en rechts ontladingen gezien die bestaan uit complexen van een piek en een langzame golf, met een frequentie van 3 per seconde. Deze typische absences moeten worden onderscheiden van andere, die voorkomen in het kader van hersenziekten en gepaard gaan met tragere piek-golven.
Absence epilepsie in de kinderjaren manifesteert zich vanaf de leeftijd van 5-7 jaar en reageert veelal goed op behandeling. Tonisch-klonische aanvallen komen voor bij een minderheid van de kinderen (ongeveer 10 - 15%) (Callenbach et al., 2009; Guerrini, 2006).
In de adolescentie
De aanvallen onderscheiden zich van ”absence epilepsie in de kinderjaren” door later optreden, (vanaf het 11de jaar), langere duur, en minder grote aantallen per dag dan bij de kindervorm. Ze doen zich vooral voor bij het wakker worden. Automatismen, bijvoorbeeld van de handen, doen zich tijdens absences meer voor dan bij de kindervorm. Myoklonieën zijn niet uitzonderlijk. Meer kinderen met juveniele absences dan met absences in de kinderjaren hebben tevens gegeneraliseerde aanvallen met tonisch-klonische trekkingen. Er zijn overgangsvormen van juveniele absence epilepsie en juveniele myoclonus epilepsie (zie Ziektebeschrijving Epilepsie, epilepsiesyndromen, (Guerrini, 2006).
Medische behandeling en beloop
Medische behandeling en beloopDe diagnose wordt gesteld op grond van het verhaal van kind en ouders, en eventueel de leerkrachten, aangevuld met bevindingen bij eeg-onderzoek.Dubbelblind, gerandomiseerd en gecontroleerd) onderzoek heeft nog weinig plaats gevonden. Behandeling van kinderen met absence epilepsie kan het beste beginnen met ethosuxemide. Valproaat is in gelijke mate effectief maar heeft meer nadelig effect op de aandacht van kinderen dan ethosuximide. Lamotrigine is minder effectief dan de twee genoemde middelen. Het nadelig effect op de aandacht is ongeveer gelijk aan dat van ethosuximide (Glauser et al. 2010). Deze bevindingen behoeven wel nog bevestiging. Niet zelden is behandeling met twee in plaats van een van de drie middelen nodig (zie ook Guerrini, 2006). Kinderen die alleen absences hebben reageren goed op behandeling en worden in de regel aanvalsvrij. Veel kinderen met absences en gegeneraliseerde tonisch-clonische trekkingen hebben langdurig behandeling met anti-epileptica nodig (Callenbach et al., 2009).
Cognitief functioneren
Cognitief functionerenAlgemene intelligentie
Kinderen met absence epilepsie hebben groepsgewijze vaak een wat lager intelligentiequotiënt (IQ) dan gezonde leeftijdgenoten die ter controle worden onderzocht. Het verschil betreft het totale IQ (TIQ) en de beide componenten verbaal IQ (VIQ) en performaal IQ (PIQ). Ter illustratie: het totale IQ van 16 rechtshandige kinderen, die sinds zij medicatie gebruikten geen of weinig absences hadden, varieerde van 71 tot 120, mediaan 91; van de controlegroep varieerde het totale IQ van 91-119, mediaan 103 (Pavone et al., 2001). In een ander onderzoek bedroeg het totale IQ van 69 kinderen met absence epilepsie gemiddeld 100 en van de controlegroep 111 (Caplan et al., 2008). Maar direct na diagnose (voor instelling op anti-epileptica) en een jaar later verschilden een kleine groep kinderen met absence epilepsie niet van klasgenootjes (IQ’s 99 en 101) (Schouten & Oostrom, 2001). Deze groep was medicamenteus goed behandelbaar. Dagelijks grote aantallen absences kunnen kinderen belemmeren in waarnemen en handelen.
Andere cognitieve functies
Kinderen met absences scoren in verschillende domeinen gemiddeld net wat lager dan controlekinderen: volhouden van aandacht, inprenten en onthouden van woorden, opsommen van woorden die behoren tot bepaalde betekenisvelden(bijvoorbeeld: groente of vervoermiddel) of van woorden die beginnen met bepaalde letters lukken bij kinderen met absence epilepsie gemiddeld wat minder dan bij normale leeftijdsgenootjes (Henkin et al. 2005). Een lichte achterstand in cognitief functioneren bestaat al kort nadat de absences zijn gediagnosticeerd; ze lijkt een verschijnsel te zijn van de aandoening, die ook de absences veroorzaakt (Bhise et al., 2009; Mandelbaum et al., 2009). Maar bij herhaling van het onderzoek blijken telkens andere kinderen de lagere scores te behalen. Het iets mindere presteren zou dus ook nog een uiting kunnen zijn van onzekerheid of andere niet-biologische invloeden (Oostrom et al., 2005). Daar staat weer tegenover dat veel brusjes van kinderen met absence epilepsie ook ietwat lager scoren dan de groep waarop de tests genormeerd werden. Dit verschijnsel verklaart men wel uit gemeenschappelijke genetische invloeden, maar het is even goed mogelijk dat ook hier niet-biologische factoren belangrijker zijn.
Schoolloopbaan
De meeste kinderen met absence epilepsie volgen gewoon basis onderwijs. Maar de percentages kinderen die een groep doubleren en/of extra onderwijskundige ondersteuning nodig hebben zijn groter dan de landelijke percentages (Oostrom et al., 2002; Wirrell et al, 1997).
Psychosociaal
PsychosociaalDe absence epilepsieën behoren tot de gunstigste vormen van epilepsie, maar helemaal zonder gevolgen blijven ze niet. Zoals blijkt uit vragenlijsten over het emotionele, psychosociale en lichamelijke functioneren van kinderen met absence epilepsie, vinden ouders dat emotionele of gedragsproblemen hun kinderen met nog actieve absence epilepsie meer belemmeren in hun schoolwerk of andere activiteiten dan gezonde kinderen. Ze vinden hun kinderen ook opvliegender en minder zeker van zichzelf. Ze zien de gezondheid van hun kinderen met absence epilepsie als kwetsbaarder dan ouders kinderen zonder epilepsie vinden (Jennekens-Schinkel et al., 2004). Andere belangrijke informatie van ouders is dat kinderen met absence epilepsie voor ze in slaap vallen langer wakker liggen dan gezonde kinderen, dat ouders hen vaker ’s nachts moeten geruststellen en dat ze de kinderen overdag slaperiger vinden dan gezonde kinderen. Ouders klagen bij uitzondering over aandachtstekort met overmatige bewegelijkheid (Attention Deficit and Hyperactivity Disorder, ADHD) (ENCYCL-ADHD). De kinderen zelf, vooral de meisjes, worden op den duur bang voor de aanvallen; ze lijken hoe langer hoe meer het gevoel te krijgen dat ze geen controle hebben (Caplan et al., 2008).
Gevolgen voor ouders en gezin
De ouders van kinderen met nog actieve absence epilepsie hebben veel zorgen om hun kind. Maar ook al heeft het kind nog aanvallen, absence epilepsie bepaalt de gezinsactiviteiten nauwelijks meer dan andere gebeurtenissen en ze veroorzaakt zelden extra spanning in het gezin (Jennekens-Schinkel et al., 2004).
Psychosociale toekomst
Over de psychosociale gevolgen op volwassen leeftijd is niet veel bekend, misschien doordat absence epilepsie algemeen als goedaardig wordt beschouwd. Voor zover beschikbaar, wijzen gegevens op meer gedragsproblemen, minder hoge opleiding en minder goede inpassing in de arbeidsmarkt dan van mensen die vroeger een andere chronische ziekte hadden, zoals juveniele reumatoïde artritis. De nadelige gevolgen zijn vooral duidelijk bij mensen die aanvallen houden (Wirrell et al., 1997).
Begeleiding
Begeleiding1. Ouders en leerkrachten merken absences bij kinderen vaak niet of onvoldoende op. Ook deskundigen moeten een list toepassen (laten tellen bij diep ademen) om absences uit te lokken, zorgvuldig te observeren en tijdens eeg-onderzoek te registreren. Toch kunnen de absences gevolgen hebben voor het dagelijkse functioneren: sommige kinderen zijn of worden onoplettend, drukker, of gaandeweg onzeker. Het kind heeft in de fase van actieve absences extra begrip, tolerantie en ondersteuning nodig.
2. Over het ziektebeeld in het algemeen en de verwachtingen voor de toekomst is goede aanvullende informatie beschikbaar voor ouders en leerkrachten. Ouders en betrokkenen van buiten het gezin hoeven zich in de omgang met het kind niet onzeker te voelen. Ouders en andere betrokkenen kunnen ervan uitgaan dat ook een kind met absences gewoon opgevoed kan, zelfs moet, worden.
3. Zo nodig kan de huidige situatie van dit speciale kind verhelderd worden door middel van (neuro)psychologisch onderzoek. Ten aanzien van vragen rond het schoolse leren kan beroep gedaan worden op voorlichting door landelijke epilepsie organisaties en op analyse en eventueel begeleiding door de expertisecentra voor onderwijs en epilepsie. Zie Ziektebeschrijving Epilepsie Andere teksten op internet , en Ziektebeschrijving Epilepsie, Begeleiding (punt 3).
4. De behandelend arts adviseert over extra risico’s van letsel door absence aanvallen, bijvoorbeeld in het verkeer en bij sport en spel; zo nodig lichten andere begeleiders de adviezen nader toe.
Een kind met absence epilepsie
Een kind met absence epilepsie fajenn 19 mei, 2010 - 10:11Literatuur
LiteratuurBhise VV, Burack GD, Mandelbaum DE (2009) Baseline cognition, behavior and motor skills in children with new-onset idiopathic epilepsy. Developmental Medicine & Child Neurology Augustus 21; DOI 10 1111/j 1469-8749 2009 03404.x
Callenbach PMC, Bouma PAD, Geerts AT, Arts WFM, Stroink H, Peeters EAJ, van Donselaar CA, Peters ACB, Brouwer OF (2009) Long-term outcome of childhood absence epilepsy: Dutch study of epilepsy in childhood. Epilepsy Research 83: 249-256
Caplan R, Siddarth P, Stahl L, Lanphier E, Vona P, Gurbani S, Koh S, Sankar R, Shields WD (2008) Childhood absence epilepsy: behavioral, cognitive, and linguistic comorbidities. Epilepsia 49: 1838-1846
Chioza BA, Aicardi J, Aschauer H, Brouwer O, Callenbach P, Covanis A and 17 other co-authors (2009) Genome wide high density SNP-based linkage analysis of childhood absence epilepsy identifies a susceptibility locus on chromosome 3p23-p14. Epilepsy Research 87: 247-255
Durá-Travé T, Yoldi-Petri ME, Gallinas-Victoriano F (2008) Incidence of epilepsies and epileptic syndromes among children in Navarre, Spain: 2002-through 2005. Journal of Child Neurology 23: 878-882
Glauser TA, Cnaan A, Shinnar S, Hirtz DG, Dlugos D, Masur D en 3 anderen (2010) Ethosuxemide, Valproic acid, and Lamotrigine in Childhood Absence Epilepsy. New England Journal of Medicine 362: 790-799
Guerrini R (2006) Epilepsy in children. Lancet 367: 499-524
Henkin Y, Sadeh M, Kivity S, Shabtai E, Kishnon-Rabin L, Gadoth N (2005) Cognitive function in idiopathic generalized epilepsy of childhood. Developmental Medicine & Child Neurology 47: 126-132
Jennekens-Schinkel A, Jennekens FGI (2008) Neuropsychologie van neurologische ziekten in de kindertijd. Amsterdam: Uitgeverij Boom, 420-422
Jennekens-Schinkel A, Raat H, Kool H, Knol J, Peters ACB (2004) Sleep problems, school career and health perceptions of children with rolandic or absence epilepsy. UMC Utrecht, niet gepubliceerd onderzoek.
Mandelbaum DE, Burack GD, Bhise VV ( 2009) Impact of antiepileptic drugs on cognition, behavior and motor skills in children with new-onset idiopathic epilepsy. Epilepsy & Behavior 16: 341-344
Larsson K, Eeg-Olofsson O (2006) A population based study of epilepsy in children from a Swedish county. European Journal of Neurology 10: 107-113
Oostrom KJ, Teeseling H, Smeets-Schouten A, Peters AC, Jennekens-Schinkel A (2005) Three to four years after diagnosis: cognition and behavior in children. A prospective and controlled study. Brain 128: 1546-1556
Panayiotopoulos CP (2008) Controversal topics in epilepsy. Epilepsia 49” 2131-2147
Pavone P, Bianchini R, Trifiletti RR, Incorpora G, Pavone A, Parano E (2001) Neuropsychological assessment in children with absence epilepsy. Neurology 56: 1047-1051
Wirrell EC, Camfield CS, Camfield PR, Dooley JM, Gordon KE (1997) Long-term psychosocial outcome in typical absence epilepsy. Archives of Pediatrics and Adolescent Medicine 151: 152-158
Rolandische epilepsie
Rolandische epilepsie fajenn 6 mei, 2010 - 09:43Inleiding
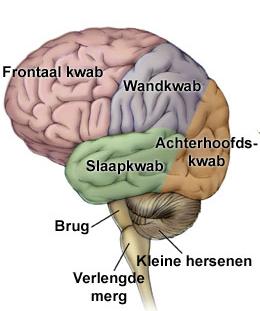
InleidingDe sulcus centralis cerebri is een groeve in het oppervlak van de grote hersenen die de grens vormt tussen het voorste, frontale deel van de grote hersenen en het daar achterliggende pariëtale deel. De sulcus is zowel links als rechts aanwezig. Hij wordt ook wel fissura (spleet) Rolandiï genoemd naar de arts-anatoom Luigi Rolando die op de overgang van de 18de naar de 19de eeuw leefde in Noord Italië. Zenuwcellen in de hersenschors direct voor de groeve hebben een sturende functie in de motoriek, zenuwcellen direct achter de groeve zijn essentieel voor gevoelswaarnemingen (sensibiliteit).
Kinderen met Rolandische epilepsie (RE) hebben kortdurende aanvallen van sensibele en motorische verschijnselen aan één kant van gelaat, mondholte en keel, al of niet samen met overeenkomstige verschijnselen in de gelijkzijdige hand of in gelijkzijdige hand, arm en been. Op het eeg worden laag in het Rolandische gebied tot in de temporale kwab complexen gezien van hoge piekgolven gevolgd door een langzame golf. RE wordt ook wel aangeduid als benigne epilepsie met centrotemporale pieken bij kinderen, of “benign childhood epilepsy with centrotemporal spikes”, afgekort BCECTS. In deze tekst worden eerst de lichamelijke verschijnselen kort samengevat. Daarna wordt ingegaan op de gevolgen van deze vorm van epilepsie voor cognitie en gedrag.
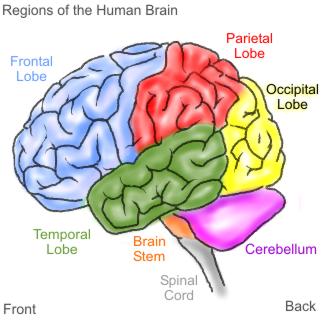
Figuur overgenomen via Google afbeeldingen
Figuur overgenomen via Google afbeeldingen
Andere teksten op internet
Andere teksten op internetwww.kinderneurologie.eu biedt deskundige Nederlandstalige informatie voor familieleden en hulpverleners.
www.ilae-epilepsy.org/ctf/ is de website van het internationale verbond tegen epilepsie. Twee vooraanstaande auteurs bieden Engelstalige informatie voor (para)medici. Zie bij epilepsie syndromen.
Voor websites over informatie aan familieleden, contact tussen familieleden en ondersteuning en advies bij schoolproblemen, zie Ziektebeschrijving Epilepsie Andere teksten op internet.
Epidemiologie
EpidemiologieVan de schoolkinderen met epilepsie heeft 15 tot 25% RE; de aandoening komt iets meer voor bij jongens dan bij meisjes (Høie et al., 2008; Nicolai et al., 2005) . RE begint ergens tussen de leeftijden van 2 en 13 jaar, met een piek op de leeftijd van 7-8 jaar. Ze verdwijnt omstreeks het 15de jaar of eerder. De kans op familiair voorkomen is verhoogd (Bali et al., 2007).
Oorzaken
OorzakenStructuurafwijkingen van de hersenen zijn bij kinderen met de typische vorm van RE niet aangetoond (Fountain, 2008). Het eeg van de niet epileptische broertjes en zusjes van kinderen met RE toont in bijna 50% van de gevallen dezelfde karakteristieke centrotemporale piekgolven. Een percentage van 50 past bij autosomaal dominante overerving (Bali et al., 2007).De piekgolven worden vooral laag in het Rolandische gebied gezien, waar zich zenuwcellen bevinden die de innervatie van het gelaat regelen en in het nabije temporale gebied.
Eeg-afwijkingen die lijken op die bij RE komen voor bij het syndroom van Panayiotopoulos (zie Ziektebeschrijving Epilepsie, Epilepsiesyndromen waarbij de aanvallen vooral vegetatief zijn (misselijkheid, braken, speekselvloed, incontinentie voor urine, verwardheid, bewustzijnsdaling en trekkingen). De vegetatieve aanvallen verdwijnen na één tot twee jaar en kunnen dan plaats maken voor RE. RE gaat bij uitzondering over in het syndroom van Landau-Kleffner (LKS), een aandoening gekenmerkt door taalfunctietoornissen in combinatie met epileptische aanvallen en gedragsafwijkingen (zie Ziektebeschrijving Syndroom van Landau-Kleffner). RE kan ook bij uitzondering overgaan in een aan LKS verwant syndroom: “continue pieken en golven tijdens non- REM slaap” (Kramer, 2008). De vraag is of deze verschillende epilepsie syndromen berusten op dezelfde of verwante genetische basis.
Symptomatische epilepsie tengevolge van bijvoorbeeld corticale dysplasie, perinatale of prenatale hersenbeschadiging kan zich presenteren als RE.
Lichamelijke verschijnselen
Lichamelijke verschijnselenDe verschijnselen en eeg afwijkingen zijn niet bij alle kinderen helemaal typisch en gemakkelijk te herkennen. Zie de atypische verschijnselen.
Kenmerkende verschijnselen
De aanvallen ontstaan bij tenminste 50% van de kinderen tijdens de slaap, meestal ’s nachts, kort na het inslapen of voor het wakker worden. Veel kinderen zijn angstig als ze tijdens een aanval wakker worden. Verschijnselen van RE zijn prikkelingen aan de binnenzijde van een wang en/of één zijde van lippen of tong, kortdurend onvermogen tot spreken, en/of tot slikken waardoor speekselvloed (kwijlen), keelgeluiden, en trekkingen in wang, tong of lippen. Uitbreiding van de trekkingen naar gelijkzijdige hand, arm, been of over het gehele lichaam (zogeheten secundaire generalisatie) gaat gepaard met daling van bewustzijn. Na enkele minuten zijn de kinderen weer helemaal helder. De aanvallen duren kort (seconden of minuten), ze ontstaan bij de meeste kinderen niet vaker dan incidenteel, met tussenperiodes van 2 - 12 maanden, maar bij 20% komen ze frequent, soms zelfs dagelijks, voor (Jennekens-Schinkel & Jennekens, 2008; Guerrini, 2006).
Atypische verschijnselen
Problemen met het geven van aandacht al of niet met overbeweeglijkheid, plotseling vallen door atonie van een been, absences, therapieresistente spiertrekkingen bij vroeg begin van de aanvallen (voor het derde levensjaar), toenemende cognitieve functiestoornissen, perioden van aanhoudende zwakte aan één zijde van het gelaat met moeite bij kauwen en slikken, speekselvloed en onvoldoende articuleren: al deze verschijnselen kunnen voorkomen bij RE of in combinatie met centrotemporale pieken (Kramer, 2008).
Cognitief functioneren
Cognitief functionerenIntelligentie
In groepen kinderen met RE is het intelligentiequotiënt (IQ) gemiddeld weinig of niet lager dan in groepen controlekinderen (Høie et al., 2008; Lindgren et al., 2004; Schouten et al.,2002). Twee illustraties:
1. in onze studie van kinderen met idiopathische epilepsie vonden wij direct na diagnose bij 16 kinderen met RE een IQ van gemiddeld 99, gelijk aan het gemiddelde IQ in de gezonde controlegroep. Bijna vier jaar later was het IQ in de groep kinderen met RE gemiddeld 102 en in de controlegroep 101 (Oostrom et al., 2005; Schouten et al., 2002).
2. Ongeveer vijf jaar na het begin van de aanvallen was het totale IQ (TIQ) van 26 kinderen met RE gemiddeld 94 (standaarddeviatie 13) en van 25 “gematchte” klasgenoten 99 (standaarddeviatie 12). Een overeenkomstig klein verschil was eerder, tweeënhalf jaar na het begin van de aanvallen, gevonden (Lindgren et al., 2004).
Andere cognitieve functies
In tests van taal, geheugen en leren, (na)tekenen, aandacht en tempo zijn de gemiddelde resultaten van groepen kinderen met RE vaak net iets lager dan van controlekinderen of dan het gemiddelde van de hele leeftijdsgroep in de bevolking. Waar de ene studie afwijkingen vindt in bepaalde domeinen, zoals taal- en/of geheugen, zijn in andere studies andere domeinen zwak. Mogelijk werkt een algemenere factor zoals gebrekkige aandacht of een laag tempo door in veel verschillende taken. Ter illustratie: Kinderen met RE hadden vergeleken met gezonde kinderen meer moeite om hun aandacht op iets te richten wanneer de geboden tijd heel kort was, maar ze functioneerden normaal als er iets meer verwerkingstijd was. Ze hadden vooral last van tijdtekort als ze door een verkeerd waarschuwingssignaal een prikkel verwachtten op verkeerde plekken van een beeldscherm. Vooral het losmaken van de aandacht voor de ten onrechte verwachte plek kostte meer tijd (Deltour et al., 2008).
Na verloop van tijd verdwijnen de verschillen met gezonde controlekinderen merendeels (Völkl-Kernstock et al., 2009; Lindgren et al., 2004). In de volwassenheid zijn er geen cognitieve functiestoornissen of negatieve gevolgen voor opleiding of beroep.
De variabiliteit in het functioneren van kinderen met RE kan samenhangen met verschillende omstandigheden. De leeftijd is belangrijk: oudere kinderen hebben geen of nauwelijks stoornissen (Danielsson & Petermann, 2009; Lindgren et al., 2004). Op grond hiervan is wel aangenomen dat de stoornissen een uiting zijn van een rijpingsachterstand van de hersenen; met verdwijnen van de achterstand verdwijnen ook de cognitieve tekorten. Ook kunnen factoren een rol spelen zoals het meer of minder typische karakter van aanvallen, van eeg-afwijkingen, en van de frequentie en/of het tijdstip van de aanvallen en de epileptiforme ontladingen in het EEG (denk aan nachtelijke aanvallen met slaaponderbreking).
Spraak
Spraak, spraakwaarneming en auditief verwerken zijn mede afhankelijk van processen in het Rolandische gebied. Het zou dus kunnen dat kinderen met RE hier moeite mee hebben. Lang niet alle kinderen met RE, maar wel meer dan gezonde kinderen, laten in de fase van het leren praten spraakklanken weg of vervangen de juiste door verkeerde spraakklanken en zijn daardoor slecht of matig verstaanbaar (Clarke et al., 2007). Meestal gaat het om medeklinkers. In het begin van de basisschoolleeftijd hebben meer kinderen met RE ook moeite met “hakken-en-plakken” (ENCYCL-Taalverwerving), maar bij kinderen van gemiddeld 9 jaar is het onderscheiden van spraakklanken zowel in het spreken als in het verstaan normaal (Northcott et al., 2007; Chaix et al., 2005).
Schoolvaardigheden
Leren lezen, schrijven en rekenen is voor aanzienlijk meer kinderen met RE dan voor gezonde controlekinderen moeilijk. Kinderen die moeite hebben met rekenen leren ook moeilijk lezen en schrijven. Zijn de basisvaardigheden eenmaal verworven, dan valt het voor kinderen met RE verder wel mee.
Jongere leeftijd waarop de aanvallen verschijnen en meer eeg-afwijkingen gedurende langere perioden lijken de kans op ontstaan van de genoemde leerproblemen te vergroten (Piccinelli et al., 2008; Papavasiliou et al., 2005). Meer kinderen met RE dan gezonde kinderen hebben extra hulp nodig. Voor kinderen met verschillende typen idiopathische epilepsie, waaronder 15 kinderen met RE) is aangetoond dat door de jaren heen telkens andere kinderen zwakker dan de controlekinderen presteren en dat de kinderen met RE niet behoorden tot degenen die kampten met blijvende leerproblemen (Oostrom et al., 2005; Schouten & Oostrom, 2001).
Gedrag
Gedragsproblemen komen bij kinderen met RE in wisselende mate voor in de actieve fase van de ziekte maar als er geen aanvallen meer zijn, verdwijnen meestal ook de gedragsproblemen (Völkl-Kernstock et al. , 2009; Nicolai et al., 2005). Er zijn ook kinderen met RE over wie de ouders of leerkrachten altijd normaal gedrag melden (Lindgren et al., 2004).
Begeleiding
BegeleidingBegeleiding
• Een correct gestelde syndroom diagnose “Rolandische epilepsie” heeft tot groot voordeel dat ouders en kind geïnformeerd kunnen worden over het doorgaans niet ernstige en voorbijgaande karakter van de aandoening. Voor neuropsychologische diagnostiek is in de regel geen aanleiding.
• De aanvallen komen bij de meeste kinderen sporadisch voor waardoor ze niet altijd nopen tot medicamenteuze behandeling. Anti-epileptica verschillen in cognitieve bijwerkingen: lamotrigine bijvoorbeeld heeft weinig negatief effect op cognitieve functies, duidelijk minder dan carbamazepine (zie Ziektebeschrijving Epilepsie. Behandeling. Antiepileptica
• De aanvallen komen bij ongeveer de helft van de kinderen alleen ‘s nachts voor. Sommige kinderen zijn daardoor bang naar bed te gaan. Het risico dat omstanders een aanval zien, daarvan schrikken en daardoor het kind met RE anders bejegenen – met de kans op stigmatisering – is bij nachtelijke aanvallen gering. Wel bestaat het gevaar dat de slaap onvoldoende verkwikkend is en dat de kinderen daardoor overdag minder alert zijn en/of aandachtsproblemen hebben.
• Het risico dat de Rolandische epilepsie bij het ouder worden van het kind overgaat in een andere vorm van epilepsie is klein maar aanwezig. De ouders behoeven daarover informatie. Bij tekenen van een andere vorm van epilepsie wordt klinisch neuropsychologisch onderzoek aangeraden mede om later eventuele progressie van afwijkingen te kunnen vaststellen.
Literatuur
LiteratuurBali B, Kull LL, Strug LJ, Clarke T, Murphy PL, Akman CI, Greenberg DA, Pal DK (2007) Autosomal dominant inheritance of centrotemporal sharp waves in rolandic epilepsy families. Epilepsia 48: 2266-2272
Chaix Y, Laguitton V, Lauwers-Cancès V, Daquin G, Cancès C, Démonet J-F, Villeneuve N (2006) Reading abilities and cognitive functions of children with epilepsy: influence of epilepsy syndrome. Brain & Development 28: 122-130
Clarke T, Strug LJ, Murphy PL, Bali B, Carvalho J, Foster S, Tremont G, Gagnon BR, Dorta N, Pal DK (2007) High risk of reading disability and speech sound disorder in rolandic epilepsy families: case-control study. Epilepsia 48: 2258-2265
Deltour L, Querné L, Vernier-Hauvette M-P, Berquin P (2008) Deficit of endogenous spatial orienting of attention in children with benign epilepsy with centrotemporal spikes (BECTS). Epilepsy Research 79: 112-119.
Fountain NB (2008) Evidence for functional impairment but not structural disease in benign rolandic epilepsy. Epilepsy Currents 8: 14-
Guerrini R (2006) Epilepsy in children. Lancet 367: 499-524
Høie B, Sommerfelt K, Waaler PE, Alsaker FD, Skeidsvoll H, Mykletun A (2008) The combined burden of cognitive, executive function, and psychosocial problems in children with epilepsy: a population-based study. Developmental Medicine & Child Neurology 50: 530-536
Jennekens-Schinkel A, Jennekens FGI (2008) Neuropsychologie van neurologische aandoeningen in de kindertijd. Amsterdam, Uitgeverij Boom, pp 417-420
Kramer U (2008) Atypical presentations of benign childhood epilepsy with centrotemporal spikes: a review. Journal of Child Neurology 23: 785-790
Lindgren A, Kihlgren M, Melin L, Croona C, Lundberg S, Eeg-Olofsson O (2004) Development of cognitive functions in children with rolandic epilepsy. Epilepsy & Behavior 5: 903 - 910
Nicolai J, Aldenkamp AP, Arends J, Weber JW, Vles JSH (2006) Cognitive and behavioral effects of nocturnal epileptiform discharges in children with benign childhood epilepsy with centrotemporal spikes. Epilepsy & Behavior 12: 494-496
NorthcottE, Connolly AM, Berroya A, McIntyre J, Christie J, Taylor A, Bleasel AF, Lawson JA, Bye AME (2007) Memory and phonological awareness in children with benign rolandic epilepsy compared to a matched control group. Epilepsy Research 75: 57-62
Oostrom KJ, van Teeseling H, Smeets-Schouten A, Peters ACB, Jennekens-Schinkel A on behalf of the Dutch Study of Epilepsy in Childhood (2005) Three to four years after diagnosis: cognition and behaviour in children with ‘epilepsy only’. Brain 128: 1546-1555
Papavasiliou A, Mattheou D, Bazigou H, Kotsalis C, Paraskevoulakos E (2005) Written language skills in children with benign epilepsy with centrotemporal spikes. Epilepsy & Behavior 6: 50-58
Piccinelli P, Borgatti R, Aldini A, Bindelli D, Ferri M, Perna S, Pitillo G, Termine C, Zambonin F (2008) Academic performance in children with rolandic epilepsy. Developmental Medicine & Child Neurology 50: 353-356.
Schouten A, Oostrom KJ, Pestman WR, Peters ACB, Jennekens-Schinkel A (2002) Leaarning and memory of school children with epilepsy: a prospective controlled longitudinal study. Developmental Medicine & Child Neurology 44: 803-811
Schouten A , Oostrom KJ, Peters ACB, Pestman WR, Jennekens-Schinkel A (2001) The risk of disappointing school results of children with idiopathic or cryptogenic epilepsy antedates diagnosis – a study of errors and peculiarities of execution in school tasks. In: Cognition and Behaviour of schoolchildren with newly diagnosed idiopathic or cryptogenic epilepsy. Thesis: Universiteit Utrecht, Ch 4, pp. pp 41-56.
Völkl-Kernstock S, Bauch-Prater S, Ponocny-Seliger E, Feucht M (2009) Speech and school performance in children with benign partial epilepsy with centro-temporal spikes (BCECTS). Seizure 18: 320-326
Temporale epilepsie
Temporale epilepsie fajenn 19 mei, 2010 - 11:50Inleiding
InleidingPlaatselijke (focale) overmatige ontlading van zenuwcellen in de temporale gebieden van de hersenen kan plotseling kortstondige abnormale verschijnselen (aanvallen) veroorzaken. De verschijnselen kunnen bestaan uit doelloze bewegingen of vreemde zintuiglijke ervaringen. De aanvallen worden “partieel” genoemd als ze niet uitbreiden tot trekkingen over het halve of hele lichaam. Breiden ze wel uit over het lichaam, dan benoemt men dat als secundaire generalisatie. De verschijnselen heten “complex partieel” als er bewustzijnsdaling bij optreedt.
We vatten de oorzaken en de aanvalsverschijnselen van temporale epilepsie samen en gaan daarna nader in op cognitieve en gedragsstoornissen.
Figuur overgenomen via Google afbeeldingen van www.skep.be
Figuur overgenomen via Google afbeeldingen van www.evodisku.multiply.com
Andere teksten op internet
Andere teksten op internetVoor informatie aan ouders en andere familieleden, voor het leggen van contacten tussen familieleden en voor advies en ondersteuning bij schoolproblemen, zie Ziektebeschrijving Epilepsie, Andere teksten op internet.
Epidemiologie
EpidemiologieIn 2005 waren er in Nederland iets minder dan vier miljoen kinderen. Volgens gegevens van huisartsen hadden ruim 9.000 daarvan epilepsie (Jennekens-Schinkel & Jennekens, 2008). Bijna de helft van deze kinderen heeft focale epilepsie; van de focale epilepsieën is een grote minderheid temporaal (Guerrini, 2006).
Oorzaken
OorzakenIn ongeveer 20% van de gevallen wordt geen duidelijke oorzaak gevonden. De epilepsie is dan idiopathisch. Genetische factoren zouden in deze gevallen een rol kunnen spelen. Bij de idiopathische vorm hebben veelal familieleden epilepsie of koortsstuipen (gehad). Er is ook een autosomaal dominant overervende temporale epilepsie (“autosomal dominant partial epilepsy with auditory features”, ADPEAF). In ongeveer 80% van de gevallen is de epilepsie symptomathisch. De temporale kwab kan door afwijkende ontwikkeling misvormd (dysplastisch) zijn, hij kan een nauwelijks groeiende tumor of een abnormale vaatstructuur bevatten. Bij een aanmerkelijke minderheid (ongeveer 20-30%) van de kinderen met temporale epilepsie is het mediale deel van de temporale kwab, dat wil zeggen de hippocampus en nabije structuren, sclerotisch. De hippocampus is een licht gekromd langwerpig gebied dat aan de mediale kant uitpuilt in het temporale deel van de ventrikel. Het percentage zogeheten mesotemporale sclerose (MTS) of hippocampussclerose is het kleinst bij zeer jonge kinderen met temporale epilepsie, het benadert bij schoolkinderen 50 maar is dan nog altijd kleiner dan bij volwassenen (Jennekens-Schinkel & Jennekens, 2008; Fontana et al., 2007). Hoe de sclerose ontstaat is niet helemaal zeker; ze zou een gevolg kunnen zijn van langdurige koortsstuipen in een eerdere fase van de ontwikkeling (Provenzale et al., 2008). Dysplasie en sclerose komen vaak gecombineerd voor. (Tassi et al., 2009; Provenzale et al., 2008).
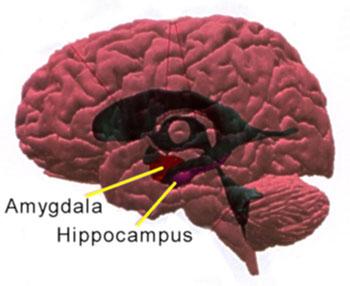
In de temporale hoorn van het ventrikelsysteem zijn de locaties van de hippocampus en de nabij gelegen amandelkern aangegeven. Deze figuur is afkomstig van huehueteotl.wordpress.com en overgenomen via Google afbeeldingen
Aanvalsverschijnselen
AanvalsverschijnselenFunctieverdeling in de temporale gebieden bepaalt de aard van de aanvalsverschijnselen
Functies van de hersenen zijn in netwerken georganiseerd; de netwerken kunnen zichtbaar gemaakt worden met behulp van bijvoorbeeld functionele MRI (fMRI). Hersengebieden die in het bijzonder betrokken zijn bij een bepaalde functie zijn in zo’n netwerk opgenomen. In de temporale kwab onderscheidt men een fylogenetisch oud mediaal deel van het fylogenetisch jongere laterale. De hippocampus en andere, kort bij de hippocampus gelegen structuren behoren tot het oude deel. De hippocampus heeft een bijzondere taak in geheugenfuncties. In het laterale deel van de temporale kwab bevinden zich gebieden met een bijzondere betekenis voor het begrijpen van taal (centrum van Wernicke) en van geluiden. De eerste verschijnselen van een focale aanval weerspiegelen de functie van het gebied waar de excessieve ontladingen ontstaan.
Verschijnselen
De eerste aanvallen ontstaan bij de meeste kinderen voor het zesde levensjaar, zelfs al in de eerste levensmaanden. Maar het debuut kan ook op schoolleeftijd plaatsgrijpen. Bij kinderen met MTS debuteert de epilepsie meestal pas in het 5de tot 6de levensjaar of later. Het aantal aanvallen kan variëren van meerdere per dag tot enkele per maand of minder (Fontana et al., 2007; Ray & Kotagal, 2005) .
- Bij kinderen van 6-7 jaar en ouder zijn de verschijnselen zoals bij volwassenen. De aanval begint meestal met een aura: bijvoorbeeld een opstijgend onaangenaam gevoel in de maagstreek, tegen beter weten in menen iets wat men nu waarneemt al eens eerder precies zo te hebben gezien (déjà-vu) of gehoord (déjà-entendu), een bepaalde smaak of reukgewaarwording, maar ook wel een intens gevoel van emotie of een zintuiglijke illusie (bijvoorbeeld macropsie of micropsie). Hier kan het bij blijven, maar veelal volgen andere verschijnselen: onderbreking van bezigheid, starende blik, wijd open ogen, wijde pupil, motorische automatismen zoals smakken, likken van de lippen, kauwen, friemelen met de handen, plukken aan de kleren, open- en dichtdraaien van een kraan, abnormale stand (dystoon) van hand of ander lichaamsdeel aan de lichaamszijde die gekruist is met de plaats van het focus.
- Bij kinderen jonger dan 6-7 jaar komen aura en kortdurend contactverlies minder voor, of zijn moeilijker registreerbaar dan bij oudere kinderen. Automatismen zijn er wel maar ze zijn eenvoudig. Motorische verschijnselen zoals tonische contracties en spiertrekkingen (myoklonieën) staan meer op de voorgrond en kunnen tweezijdig voorkomen.
Bij secundaire generalisatie ontstaat een tonische contractie en volgen daarna klonische krampen.
Medicamenteuze behandeling
Medicamenteuze behandelingLamotrigine en carbamazepine behoren volgens een niet-geblindeerde, gerandomiseerde en gecontroleerde studie tot de effectiefste middelen voor de behandeling van focale epilepsie (Marson et al., 2007; zie ook Bouma, 2009, en de Richtlijn Epilepsie). Lamotrigine wordt geacht verhoudingsgewijze weinig effecten te hebben op de cognitie (Meador, 2008; zie ook Ziektebeschrijving Epilepsie, Behandeling). De anti-epilepsie medicatie werkt het best bij de idiopathische vorm van temporale epilepsie. Sommige kinderen reageren onvoldoende op medicatie; bij die kinderen moet de mogelijkheid van operatieve behandeling worden uitgezocht (Guerrini, 2006).
Cognitie en gedrag
Cognitie en gedragIntelligentie
Kinderen met medicamenteus goed behandelbare temporale epilepsie hebben in de regel een normale intelligentie, althans de eerste jaren na diagnose (Oostrom et al., 2003). Wanneer het goed blijft gaan met de aanvallen blijft de intelligentie op peil.
Van de kinderen met medicamenteus niet behandelbare temporale epilepsie die in aanmerking komen voor epilepsiechirurgie heeft bijna de helft een zwakke intelligentie (IQ < 80) of een verstandelijke beperking (IQ < 70), die voor zover tot nu toe bekend bij 10–15% van de kinderen matig tot zeer ernstig is (IQ < 50) (De Koning et al., 2009).
Waardoor een plaatselijke afwijking in een deel van een temporale kwab bij kinderen zo grote gevolgen heeft voor de intelligentie is onduidelijk. De aard van de afwijking en in welke hersenhelft de epilepsie ontstaat, lijken niet bepalend. Aannemelijk is dat de leeftijd bij ontstaan van de aanvallen een rol speelt; klaarblijkelijk zijn de epileptische aanvallen voor de ontwikkeling van de hersenschors van jonge kinderen ongunstig (Cormack et al., 2007). Op volwassen leeftijd ontstane temporale epilepsie verstoort veeleer het geheugen dan de intelligentie.
Leren en Geheugen
Heel verschillende dingen – zoals woorden, verhalen, wegen, plattegronden, muziek en emoties – worden geleerd, onthouden en vaak ook vergeten. Bij dat alles zijn de temporale hersengebieden betrokken. Het volgende is bekend over leren en geheugen van kinderen met temporale epilepsie.
- Jonge kinderen met temporale epilepsie leren en onthouden lijsten woorden in het algemeen niet minder goed dan gezonde leeftijdgenootjes (ENCYCL-Geheugen). Maar in de tweede helft van de schoolleeftijd verbetert hun leer- en geheugencapaciteit wat minder dan van de gezonde kinderen, vermoedelijk doordat de kinderen hun hele handelen, met inbegrip van kennisverwerving, minder goed organiseren. Jongeren met temporale epilepsie komen eerder aan op het omslagpunt van steeds beter naar geleidelijk minder goed leren en onthouden (adolescenten met temporale epilepsie een vijftal jaren eerder dan gezonde personen) (Helmstaedter & Elger, 2009). Een focus in de linker temporale kwab is na de kinderjaren voor het leren van taalmateriaal ongunstiger dan een focus in de rechter temporale kwab. Hippocampussclerose is door alle leeftijden heen een negatieve factor voor leren en onthouden, doordat de geheugen”sporen” minder sterk zijn. (Helmstaedter & Elger, 2009). Over leren en onthouden van informatie die niet in taal is vervat is minder bekend dan over talige informatie.
- Verhalen met een emotionele lading worden in het algemeen beter opgenomen en onthouden dan neutrale. Kinderen met vroeg ontstane moeilijk behandelbare temporale epilepsie ondervinden mogelijk deze invloed van emotie op onthouden niet of veel minder (Jambaqué et al., 2009).
- Kinderen met al op heel jonge leeftijd epilepsie vanuit mesotemporale structuren lijken vier van vijf basisemoties minder goed te herkennen van gelaatsuitdrukkingen dan gezonde leeftijdgenootjes. Alleen blijheid lijken ze normaal af te lezen, maar bij verdriet, vrees, walging en boosheid maken ze meer fouten (Golouboff et al., 2008).
Taal
Kinderen met goed behandelbare temporale epilepsie hebben in de regel een normaal ontwikkelde taalfunctie. De meerderheid van de kinderen met moeilijk behandelbare epilepsie hebben een achterstand in de ontwikkeling van woordenschat en grammatica. Deze past meestal bij de algehele stoornis van het cognitieve functioneren. Verschijnselen van afasie hebben de kinderen niet (De Koning et al., 2009).
Andere cognitieve functies
- Ooit werd een jongen beschreven die op de leeftijd van 7 jaar in een fase van ernstige aanvallen vanuit de temporale gebieden van de rechterhersenhelft traag en zonder enige intonatie sprak terwijl woordkeuze en vervoeging en verbuiging van wat hij zei in orde waren. De aprosodie verdween toen de aanvallen verbeterden. Later ontstond de indruk dat kinderen met moeilijk behandelbare temporale epilepsie langzamer ontwikkelen dan gezonde kinderen in het begrijpen van emoties die worden overgebracht door gebaren of door de stem (Cohen et al., 1990). Over de mate van voorkomen van deze ontwikkelingsachterstand, en over de aard en de ernst ervan is nog lang geen uitsluitsel. In de paragraaf over leren en geheugen werd het tekort in de herkenning van gelaatsuitdrukkingen beschreven in termen van geheugen (voor emoties). Anderen zien de kern van het probleem als een emotietekort. Dit komt tot uiting in het doen en laten van de kinderen maar ook in hun waarnemen en begrijpen van gedrag van anderen. Het kan gelaatsuitdrukkingen en/of stemmodulatie treffen. Bij kinderen met goed behandelbare temporale epilepsie namen wij deze problemen niet waar. Het tekort zou zich niet beperken tot kinderen met een epilepsiefocus in de rechter temporale gebieden, maar bij hen wel duidelijker zijn dan bij kinderen met een linkszijdig focus.
- Kinderen met temporale epilepsie hebben moeite met natekenen van complexe figuren. Links- of rechtstemporaal focus maakt geen verschil voor de kwaliteit van het construeren (Schouten et al., 2009).
- Over het executief functioneren valt nog niet meer te zeggen dan dat kinderen met moeilijk behandelbare temporale epilepsie hun handelen minder goed organiseren (zie boven) (Rzezak et al., 2009; Jennekens-Schinkel & Jennekens, 2008) (ENCYCL-Executief functioneren).
Gedrag
Kinderen met goed behandelbare temporale epilepsie hebben niet meer gedragsproblemen dan andere kinderen van dezelfde leeftijd, tenzij er spanningsbronnen zijn in de opvoeding (ouders kunnen bijvoorbeeld hun kind met epilepsie overmatig beschermen) of in de gezinsomstandigheden. Bij kinderen met moeilijk behandelbare epilepsie kunnen gedragsproblemen samenhangen met onvoldoende herkennen van sociale signalen, zoals stembuigingen of gelaatsuitdrukkingen (Golouboff et al., 2008). Maar ze kunnen ook voortkomen uit de aard van de aandoening waar ook de epilepsie een gevolg van is of uit gebruik van bepaalde combinaties van anti-epileptica.
In uitzonderlijke gevallen kunnen psychiatrische gedragsstoornissen voorkomen, zoals paniekaanvallen en perioden van psychose met hallucinaties (“horen van stemmen” of “zien van beangstigende beelden”). Kinderen met een mentale beperking kunnen hyperactief of autistisch zijn.
Temporale epilepsiechirurgie
Temporale epilepsiechirurgieIngreep
Operatie kan worden overwogen wanneer behandeling met anti-epileptica onbevredigend blijft. Van alle vormen van epilepsiechirurgie wordt temporale resectie veruit het meest verricht. Voorafgaande aan de operatie moeten de eventuele afwijkende structuur en de aanvalsbeginzone zorgvuldig worden vastgesteld. Doel van de operatie is immers het verwijderen van het afwijkende gebied – een plaatselijke misvorming, tumor, geschrompelde hippocampus – met inbegrip van het gebied waar de epileptische aanvallen worden veroorzaakt. De meest verrichte ingreep is het verwijderen van het voorste deel van de temporale kwab, vanaf de temporale pool 3-4 centimeter achterwaarts.
Gevolgen voor aanvallen, bewegen en zien
Volgens de Landelijke Werkgroep Epilepsiechirurgie bij Kinderen is 76% van de kinderen na temporale operatie aanvalsvrij; bij de overige kinderen is het aantal aanvallen sterk verminderd (18%) of verminderd (6%) (Jennekens-Schinkel & Jennekens, 2008). In het algemeen neemt in de loop van jaren het percentage aanvalsvrije kinderen af, bij temporale epilepsie na 10 jaar tot ongeveer 50% (zowel bij kinderen als volwassenen( (Jeha et al., 2006).
De temporale kwab speelt geen rol bij de ondersteuning van motoriek en sensibiliteit en de verwijdering veroorzaakt dus geen verlammingen of gevoelsstoornissen. Wel wordt een verbinding (de lus van Meyer) van hersenstam naar het visuele deel van de occipitale schors onderbroken; hierdoor (tenzij de aandoening al tot de onderbreking geleid heeft, wat vaak voorkomt) kunnen kinderen niet zien in een kwart van het gezichtsveld van het tegenover liggende (contralaterale) oog. Door hoofd- en oogbewegingen passen kinderen zich snel bij dit tekort aan, veel sneller dan volwassenen.
Gevolgen voor cognitie
Vergeleken met de bevindingen voorafgaande aan operatie, blijft het IQ bij de meeste kinderen stabiel, ongeacht de hersenhelft die wordt geopereerd. Dat geldt zowel voor het verbale als het performale IQ. De scores van testonderzoek van het geheugen tonen geen opmerkelijke effecten van de operatie (zie voor literatuur overzicht Jennekens-Schinkel & Jennekens, 2008). De vertraagde taalontwikkeling van kinderen met medicamenteus moeilijk behandelbare temporale epilepsie verbetert niet maar verergert ook niet door temporale chirurgie. De verschillen in effecten van temporale resecties in linker en rechter hersenhelft voor de ontwikkeling van de taal zijn klein (de Koning et al., 2009). Dat is dus echt anders dan bij volwassenen.
Gevolgen voor gedrag
De verdwijning van aanvallen heeft in het algemeen een bevrijdend effect. Na temporale resecties verbeteren de kinderen in motorische activiteit en motorische vaardigheid; leven zonder aanvallen lijkt kinderen de kans te bieden om hun motorische mogelijkheden meer te ontplooien (van Empelen et al., 2005). Over andere effecten van temporale kwab operaties voor het gedrag bestaat nog onzekerheid.
Begeleiding
Begeleiding1. Globale stoornissen in het cognitieve functioneren bepalen het bestaan van veel kinderen met medicamenteus moeilijk behandelbare vormen van temporale epilepsie. De medische behandeling is gericht op tegengaan van de aanvallen en voorkomen van (verdere) cognitieve beperkingen. De begeleiding richt zich op stimuleren van ontwikkeling en aanpassen van de omgeving aan het kind; de maat hierbij wordt gevormd door het cognitieve en emotionele functioneren van het kind.
2. Zie ook de Ziektebeschrijving Epilepsie, Begeleiding.
Een kind met temporale epilepsie
Een kind met temporale epilepsie fajenn 28 mei, 2010 - 22:19Literatuur
LiteratuurCohen M, Prather A, Town P, Hynd G (1990) Neurodevelopmental differences in emotional prosody in normal children and children with left and right temporal epilepsy. Brain & Language 38: 122-134
Cormack F, Cross H, Isaacs E, Harkness W, Wright I, Vargha-Khadem F, Baldeweg T (2007) The development of intellectual abilities in pediatric temporal lobe epilepsy. Epilepsia 48: 201-204
Empelen R van, Jennekens-Schinkel A, Gorter JW, Volman MJM, Nieuwenhuizen O van, Helders PJM (2005) Epilepsy surgery does not harm motor performance of children and adolescents. Brain 128: 1536-1545
Fontana E, Negrini F, Francione S, Mai R, Osanni E, Menna E, Offredi F, Darra F, Dalla Bernardina B (2006) Temporal lobe epilepsy in children: electroclinical study of 77 cases. Epilepsia 51: 26-30
Golouboff N, Fiori N, Delalande O, Fohlen M, Dellatolas G, Jambaqué I (2008) Impaired facial expression recognition in children with temporal lobe epilepsy: impact of early seizure onset on fear recognition. Neuropsychologia 46: 1415-1428
Guerrini R (2006) Epilepsy in children. Lancet 367; 499-524
Helmstaedter C. & Elger CE (2009) Chronic temporal lobe epilepsy: a neurodevelopmental or progressively dementing disease? Brain 132: 2822-2830
Jambaqué I, Pinabiaux C, Dubouch C, Fohlen M, Bulteau C, Delalande O (2009) Verbal emotional memory in children and adolescents with temporal lobe epilepsy: a first study. Epilepsy & Behavior 16: 69-75
Jeha LE, Najm IM, Bingaman WE, Khandwala MS, Widdess-Walsh P, Morris HH, Dinner DS, Nair D, Foldvary-Schaeffer N, Prayson RA, Comair Y, O’Brien R, Bulacio J, Gupta A, Lüders HO (2006) Predictors of outcome after temporal lobectomy for the treatment of intractable epilepsy. Neurology 66: 1938-1940
Jennekens-Schinkel A, Jennekens FGI (2008) Neuropsychologie van neurologische aandoeningen in de kindertijd. Amsterdam: uitgeverij Boom. Hoofdstukken 10 en 11 over Temporale epilepsie en Epilepsie chirurgie: epidemiologie pp 390 en 406; oorzaken pp 394-395; geheugen pp 407-408; andere cognitieve functies pp 409-411; gedrag pp 411-412; temporale kwab chirurgie en gevolgen voor aanvallen pp 460-461, voor cognitie 464-465 en 470, voor gedrag 461 en 471-472.
de Koning T, Versnel H, Jennekens-Schinkel A, van Schooneveld MM, Dejonckere PH, van Rijen PC, van Nieuwenhuizen O, on behalf of the Dutch Collaborative Epilepsy Surgery Programme (DuCESP) (2009) Language development before and after temporal surgery in children with intractable epilepsy. Epilepsia 50: 2408-2419
Marson AG, Al-Kharusi AM, Appleton R, Baker GA, Chadwick DW, Cramp C, Cockerell OC, Cooper PN, Doughty J, Eaton B, Gamble C, Goulding PJ, Howell SJL, Hughes A, Jackson M, Jacoby A, Kellett M, Lawson GR, Leach JP, Nicolaides P, Roberts R, Shackley P, Shen J, Smith DF, Smith PEM, Tudor Smith C, Vanoli A, Williamson PR, on behalf of the SANAD Study Group (2007) The SANAD study of effectiveness of carbamazepine, gabapentin, lamotrigine, oxcarbazepine, or topiramate for treatment of partial epilepsy: an unblinded randomised controlled trial. Lancet 369: 1000-1015
Meador KJ (2008) Cognitive effects of levetiracetam versus topiramate. Epilepsy Currents 8: 64-65
Oostrom KJ, Smeets-Schouten A, Kruitwagen CLJJ, Peters ACB, Jennekens-Schinkel A (2003) Not only a matter of epilepsy: early problems of cognition and behavior in children with “epilepsy only” – a prospective, longitudinal, controlled study starting at diagnosis. Pediatrics 112: 1338-1344
Provenzale JM, Barboriak DP, VanLandingham K, MacFall J, Delong D, Lewis DV (2008) Hippocampal MRI signal hyperintensity after febrile status epilepticus is predictive of subsequent mesial temporal sclerosis. American Journal of Roentgenology 190: 976-983
Ray A, Kotagal P (2005) Temporal lobe epilepsy in children: overview of clinical semiology. Epileptic Disorders 7
Richtlijn Epilepsie (2006) www.neurologie.nl/richtlijnen
Rzezak P, Fuentes D, Guimarães CA, Thome-Soza S, Kuczynski E, Guerreiro M, Valente KDR (2009) Executive dysfunction in children and adolescents with temporal lobe epilepsy: is the Wisconsin Card Sorting Test enough? Epilepsy & Behavior 15: 376-381
Schouten D, Hendriksen JGM, Aldenkamp AP (2009) Performance of childrne with epilepsy on the Rey-Osterrieth complex figure test: is there an effect of localization or lateralization? Epilepsy Research 83: 184-189
Frontale epilepsie
Frontale epilepsie fajenn 19 mei, 2010 - 13:57Inleiding
InleidingPlaatselijke (focale) overmatige ontlading van zenuwcellen in de frontale kwab van de hersenen kan abnormale verschijnselen veroorzaken, zoals trekkingen of bewegingen van verschillende aard. De verschijnselen treden in aanvallen op. Ze worden partieel genoemd als ze beperkt blijven tot een deel van het lichaam. Ze worden complex genoemd als er bewustzijnsdaling bij optreedt; de bewustzijnsdaling ontstaat wanneer ontladingen zich ook voordoen buiten het oorspronkelijk frontale focus (Guerrini, 2006). Wanneer de verschijnselen zich uitbreiden over een lichaamshelft of over het gehele lichaam, dan zijn vanuit het oorspronkelijk frontale focus, zenuwcellen in de schors van de gehele gekruiste hersenhelft of van beide hersenhelften tot ontlading gekomen; de aanval is dan secundair gegeneraliseerd. We vatten de aanvalsverschijnselen van frontale epilepsie samen en gaan daarna nader in op de stoornissen van cognitie en gedrag.
Andere teksten op internet
Andere teksten op internethttp://en.wikipedia.org/wiki/Frontal_lobe_epilepsy biedt uitvoerige Engelstalige informatie over frontale kwab epilepsie, in het bijzonder over anatomie en aanvalsverschijnselen.
www.ilae-epilepsy.org/ctf is de website van het internationaal verbond tegen epilepsie met een Engelstalige medische beschrijving van nachtelijke frontaalkwab epilepsie ("autosomal dominant nocturnal frontal lobe epilepsy" of ADNFLE).
www.kinderneurologie.eu biedt een Nederlandstalige beschrijving van ADNFLE voor niet-kinderneurologen
Zie ook in deze website, de Ziektebeschrijving Epilepsie waar in de paragrafen Teksten op internet en Begeleiding website adressen worden gegeven met praktische informatie voor ouders en andere familieleden, over het leggen van contact van ouders met elkaar,en voor advies en ondersteuning bij schoolproblemen.
Epidemiologie
EpidemiologieNauwkeurige cijfers zijn niet beschikbaar. Vrij zeker is dat epilepsie bij kinderen in bijna de helft van de gevallen focaal is (Guerrini, 2006). Bij kinderen zou, anders dan bij volwassenen, extratemporale epilepsie meestal frontaal zijn en met 20 – 30% van de focale epilepsieën meer voorkomen dan de veel grondiger onderzochte temporale epilepsie.
Oorzaken
OorzakenDe meest voorkomende oorzaak van de zogeheten symptomatische frontale epilepsie van kinderen is dysplasie van de hersenschors; minder vaak wordt een langzaam groeiende tumor of een kluwen van abnormale vaten gevonden. Dysplasie blijkt vaker aantoonbaar naarmate met MRI subtielere afwijkingen zichtbaar gemaakt kunnen worden (Jeha et al., 2007).
Frontale epilepsie kan ook idiopathisch zijn. Dan is de epilepsie niet het gevolg van een andere aandoening, hetgeen in veel of alle gevallen betekent dat een genetische factor in het spel is. Autosomaal dominante nachtelijke frontale epilepsie kan door mutaties in verschillende genen worden veroorzaakt (Derry et al., 2008) .
Aanvalsverschijnselen
AanvalsverschijnselenFunctieverdeling in het frontale hersengebied
De verschillende lichaams- en gedragsfuncties van het kind worden in de hersenen ordelijk ondersteund. Die orde ontstaat dankzij verdeling van functies over functienetwerken. Deze netwerken kunnen zichtbaar worden gemaakt door bijvoorbeeld functionele (f)MRI. Bij de netwerken zijn hersengebieden betrokken die bijzondere betekenis hebben voor de op het moment van maken van de MRI actieve functie. De netwerken voor functies zoals taal veranderen met de ontwikkeling. Van belang is dat de eerste verschijnselen van een focale aanval de functie van het gebied weerspiegelen waarin zich de excessieve ontladingen voordoen.
In het grote frontale hersengebied strekt de motorische schors zich uit aan de voorzijde van de sulcus centralis die in elke hersenhelft aan de buitenkant (vlak onder de schedel) van bovenaf in benedenwaartse richting verloopt. De premotorische of supplementaire motorische schors, gelegen voor de motorische schors, draagt bij tot ondersteuning van planning en coördinatie van bewegingen. Laag in de motorische schors gelegen zenuwcellen zijn betrokken bij de motoriek van het mond-keelgebied. Direct daarvoor bevindt zich, in de supplementaire motorische schors, een kerngebied (centrum van Broca) dat bij volwassenen en bij oudere kinderen een bijzondere rol speelt bij taalverwerking. Zenuwcellen aan de mediale zijde van het frontale hersengebied, in het orbito-frontale gebied (gelegen aan de onderzijde van het frontale gebied), en in de frontale pool zijn van betekenis voor persoonlijkheidskenmerken, emoties, cognitieve functies en gedrag.
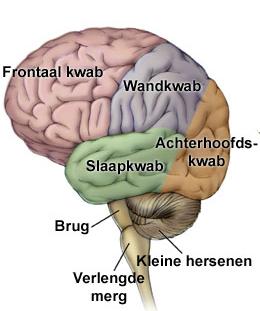
Figuur overgenomen via Google afbeeldingen van www.skep.be
De sulcus centralis bevindt zich op de overgang van de frontale kwab ("frontal lobe") en de wandkwab (pariëtale kwab).
Verschijnselen
Frontale aanvallen ontstaan plotseling, vaak in clusters (groepen), soms als in een frontale status epilepticus; de aanvallen leiden tot onderbreking van activiteit (indien overdag) en duren in de regel kort (1-2 minuten); ze komen merendeels (niet uitsluitend) ’s nachts voor; tientallen aanvallen per etmaal zijn gewoon. Na afloop van een aanval gaan de kinderen verder met hun eerdere activiteit, zijn ze niet verward, hebben ze weinig of geen klachten en beseffen ze het gebeurde niet altijd.
- De aanvallen kunnen met een aura (een gewaarwording die alleen het kind merkt), met een stemgeluid (schreeuw, bij uitzondering zingen), of met agitatie beginnen.
- Bij jonge kinderen zijn de verschijnselen vooral motorisch: spasmen, soms licht en beperkt maar soms ook massaal plotseling snel buigen of strekken van hoofd, romp, bovenarmen en benen; tonische kramp van het lichaam of een deel daarvan; klonieën van het lichaam of een deel daarvan, of myoklonieën. Automatismen komen vooral rond de mond voor.
- Bij oudere kinderen zijn de verschijnselen ook vooral motorisch, maar complexer: bewegingen die door de buitenwereld worden geïnterpreteerd als slaan of schoppen, en verder heffen van de armen, fietsbewegingen, draaibewegingen, bij een enkel kind slaapwandelen.
- Ook kunnen zich grote aanvallen met gegeneraliseerde tonische kramp gevolgd door klonische trekkingen voordoen (Guerrini, 2006; Jennekens-Schinkel & Jennekens, 2008).
Medicamenteuze behandeling
Medicamenteuze behandelingLamotrigine en carbamazepine behoren volgens een niet geblindeerde, gerandomiseerde, gecontroleerde studie tot de effectiefste middelen voor de behandeling van focale epilepsie (Marson et al., 2007; zie ook Bouma, 2009). Lamotrigine wordt geacht weinig of geen effecten te hebben op de cognitie (Meador, 2008; zie ook Ziektebeschrijving Epilepsie. Behandeling).
Cognitief functioneren en gedrag
Cognitief functioneren en gedragCognitie
- intelligentie is binnen de grenzen van het normale met dien verstande dat het intelligentiequotiënt (IQ) gemiddeld over groepen kinderen met frontale epilepsie licht verschoven is naar lagere waarden en dat het performale IQ veelal enkele punten lager is dan het verbale IQ. De intelligentie is – gemiddeld over groepen kinderen - niet zwakker dan bij andere focale vormen van epilepsie. Soms echter kan een kind dat ogenschijnlijk alert is zo oppervlakkig ingaan op de vragen en taken dat een forse verlaagd IQ wordt vastgesteld. Dit moet een signaal voor nadere epilepsiediagnostiek zijn.
- Leren van bijvoorbeeld lijsten woorden levert voor ongeveer de helft van de kinderen met frontale epilepsie onvoldoende resultaat op.
- De kinderen gaan in leer- en andere taken impulsief, onvoldoende georganiseerd en/of oppervlakkig te werk. Genoemde eigenschappen komen ook wel bij kinderen met andere vormen van focale epilepsie in wat verhoogde mate voor en zijn ook bij kinderen met frontale epilepsie niet per se blijvende kenmerken (Oostrom et al., 2005)
- Voor schrijven en tekenen is van belang dat bewegingen met twee handen of samengestelde eenhandige bewegingen star, onvoldoende behendig en traag kunnen zijn (zie voor samenvattingen van de literatuur: Patrikelis et al., 2009, Jennekens-Schinkel & Jennekens, 2008).
- Leren van schoolvaardigheden kan nadelig worden beïnvloed door veelvuldige onderbreking (hoe kort ook) van de aandacht door aanvallen.
Gedrag
Abnormaal gedrag bij zwakzinnige autistische(ENCYCL-Autisme) kinderen kan ten onrechte gedragsmatig in plaats van als symptoom van epilepsie worden begrepen.
Kinderen die regelmatig aanvallen hebben, kunnen bij uitzondering psychotisch worden waarbij ze hallucinaties hebben in de zin van het horen van stemmen of het zien van beelden die er niet zijn, ze kunnen waandenken ontwikkelen, depressief worden en bizar gedrag ontwikkelen (Sinclair & Snyder, 2008) .
Autosomaal dominante nachtelijke epilepsie uitgaande van frontale gebieden (Autosomal Dominant Nocturnal Frontal Lobe Epilepsy, ADNFLE)
Bij kinderen met ADNFLE zijn verschillende genmutaties ontdekt. Structuurafwijkingen van het hersenweefsel zijn niet aangetoond. De aanvallen ontstaan in het tweede of eerste decennium. De verschijnselen zijn zoals beschreven bij andere vormen van frontale epilepsie. Ze reageren doorgaans goed op behandeling met carbamazepine
Familiegewijze kunnen variaties in de verschijnselen voorkomen. Meestal zijn de verschijnselen vrij goedaardig, bij enkele families zijn ze ernstig. De ernst komt tot uitdrukking in het aantal nachtelijke aanvallen, het voorkomen van aanvallen overdag, de bedreigde cognitieve ontwikkeling, gedragstoornissen en onvoldoende reactie op anti-epileptica (Wood et al., 2010; Derry et al., 2008) .
Frontale epilepsiechirurgie
Frontale epilepsiechirurgieOperatie en kans op aanvalsvrijheid
Kinderen die goed medicamenteus zijn behandeld en toch aanvallen houden, en kinderen die veel nevenverschijnselen van de anti-epileptica ervaren, worden medicamenteus “onbehandelbaar” genoemd. Zij kunnen in aanmerking komen voor operatieve verwijdering van het epileptogene gebied, dat wil zeggen van het schorsgebied dat wezenlijk is voor het veroorzaken van de aanvallen. De beoordeling van de kans op goed resultaat van de ingreep is vaak moeilijk, bijvoorbeeld in het geval van dysplasie; MRI maakt deze niet altijd zichtbaar. Pas onder de microscoop kan de afwijking worden ontdekt in het uitgenomen hersenweefsel. Bovendien worden gebieden die echt nodig zijn voor het dagelijkse functioneren, zoals voor het bewegen, de taal en het geheugen, zoveel mogelijk gespaard, omdat men geen verlamming en geen afasie wil veroorzaken. Van de operaties vanwege epilepsie maken de frontale resecties een kleine minderheid uit ( 6-30%; Jeha et al., 2007). In een centrum met veel ervaring en deskundigheid op het gebied van epilepsiechirurgie constateerde men dat het percentage geopereerden dat aanvalsvrij blijft met de jaren steeds kleiner (na vijf jaar 30%, volwassenen inbegrepen) is. Maar voor de meerderheid betekent de operatie toch een aanzienlijke verbetering (Jeha et al., 2007). De kans op succes is het grootst wanneer het epileptogene gebied volledig kan worden weggenomen.
Gevolgen voor cognitie en gedrag
In het algemeen schaadt epilepsiechirurgie de cognitieve ontwikkeling van het kind niet, dat geldt ook voor frontale resecties. Operaties waarbij het centrum van Broca betrokken is zijn bij kinderen minder nadelig voor de taalfunctie dan bij volwassenen. Kennelijk kan de taalfunctie bij kinderen ook door andere gebieden in dezelfde of in de rechterhersenhelft worden ondersteund (Jennekens-Schinkel & Jennekens, 2008).
Begeleiding
Begeleiding- Evaluatie van cognitieve functies en gedrag kan gewenst zijn wanneer het kind problemen ondervindt op school. Frontale epilepsie kan zich overwegend in moeilijk gedrag uiten!
- Zie voor andere aspecten van de begeleiding de Ziektebeschrijving Epilepsie, Begeleiding in deze website.
Een kind met frontale epilepsie
Een kind met frontale epilepsie fajenn 28 mei, 2010 - 22:20Literatuur
LiteratuurBouma P (2009) Behandelingscursus in de praktijk. Effectiviteit en bijwerkingen. Basiscursus Epilepsie. www.debaer.net/sepion
Derry CP, Heron SE, Philips F, Howell S, MacMahon J, Phillips HA, Duncan JS, Mulley JC, Berkovic SF, Scheffer IE (2008) Severe autosomal dominant nocturnal frontal lobe epilepsy associated with psychiatric disorders and intellectual disability. Epilepsia 49: 2125-2129
Guerrrini R (2006) Epilepsy in children. Lancet 367: 499-524
Jeha LE, Najm I, Bingaman W, Dinner D, Widdess-Walsh P, Lüders H (2007) Surgical outcome and prognostic factors of frontal lobe epilepsy surgery. Brain 130: 574-584
Jennekens-Schinkel A, Jennekens FGI (2008) In Neuropsychologie van neurologische aandoeningen in de kindertijd. Amsterdam: Boom
- Epilepsie. Hoofdstuk 10: Aanvalsverschijnselen, cognitie en gedrag pp 400-406
- Epilepsiechirurgie. Hoofdstuk 11: Operatie in het gebied van Broca pp 454-455; gevolgen voor taal pp 473-474
Marson AG, Al-Kharusi AM, Alwaidh M (2007) The SANAD study of effectiveness of valproate, lamotrigine, or topiramate for generalised and unclassifiable epilepsy : an unblinded randomised controlled trial. Lancet 369: 1016 - 1026
Oostrom K, Teeseling H van, Smeets-Schouten A, Peters ACB, Jennekens-Schinkel ACB (2005) Three to four years after diagnosis: cognition and behaviour in children with ‘epilepsy only’, A prospective, controlled study. Brain 128: 1546-1555.
Patrikelis P, Angelakis E, Gatzonis S (2009) Neurocognitive and behavioral functioning in frontal lobe epilepsy. Epilepsy &Behavior 14: 19-26
Sinclair DB, Snyder T (2008) Psychosis with frontal lobe epilepsy responds to carbamazepine. Journal of Child Neurology 23: 431-434
Wood AG, Saling MM, Fedi M, Berkovic SF, Scheffer IE, Benjamin C, Reutens DC (2010) Neurophysiological function in patients with a single gene mutation associated with autosomal dominant nocturnal frontal lobe epilepsy. Epilepsy & Behavior 17: 531-535
Landau-Kleffnersyndroom
Landau-Kleffnersyndroom fajenn 29 mei, 2010 - 10:22Inleiding
InleidingLandau en Kleffner waren in 1957 de eersten die het naar hen genoemde syndroom (LKS) beschreven. LKS is een ziekte die ontstaat bij tot dan toe gezonde kinderen in de leeftijd van ongeveer 3 tot 8, hooguit 9 jaar (Duran et al., 2009). De taalontwikkeling van de kinderen raakt gestoord, de kinderen krijgen meestal epileptische aanvallen en er ontstaan gedragsafwijkingen. Na het 10de levensjaar volgt meestal in enige mate verbetering, van zeer gering tot vrijwel volledig. Het syndroom is zeldzaam, het veroorzaakt grote ziektelast bij de kinderen en het stelt verzorgers en behandelaars voor aanzienlijke problemen (Jennekens-Schinkel & Jennekens, 2008).
Wij vatten de verschijnselen van de aandoening samen. Daarna gaan we in op de cognitieve en gedragstoornissen en op de vraag hoe de kinderen het beste kunnen worden begeleid.
Andere teksten op het internet
Andere teksten op het internetwww.kinderneurologie.eu is een Nederlandstalige kinderneurologische website met uitvoerige informatie over epilepsie syndromen, het Landau Kleffner syndroom daarbij inbegrepen. De informatie is voor niet-medici goed toegankelijk
www.ilae-epilepsy.org/ctf is de Engelstalige website van het internationale verbond tegen epilepsie met medische informatie over het syndroom van Landau Kleffner.
Epidemiologie
EpidemiologieCijfers over het aantal kinderen dat jaarlijks LKS krijgt en over het totale aantal kinderen met LKS ontbreken. Dat geldt zowel voor Nederland als voor andere landen. In de medische literatuur zijn nu honderden kinderen gerapporteerd. De aandoening komt iets meer voor bij jongens dan bij meisjes. Voor zover bekend zijn er in Nederland tenminste enkele tientallen kinderen met dit syndroom (Nieuwenhuis & Nicolai, 2006). Het is niet uitgesloten dat lichte vormen van LKS niet gediagnosticeerd worden.
Oorzaak
OorzaakDe oorzaak van LKS is niet bekend.
- Epileptische ontladingen die vanuit de temporale kwab van de linkerhersenhelft uitbreiden naar het overeenkomstige gebied in de rechterhersenhelft zouden de taalfunctie kunnen verstoren. Het is ook mogelijk dat de epileptische ontladingen verband houden met een andere aandoening die zowel de epilepsie als de taalfunctiestoornis veroorzaakt. In elk geval zijn epileptische ontladingen geregistreerd in een gebied dat betrokken is bij woordherkenning (Castillo et al., 2008).
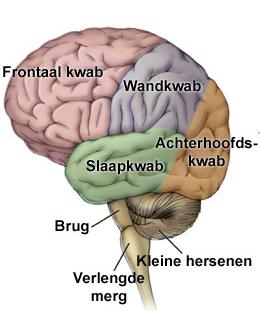
Figuur 1.Hersenoppervlak
Figuur overgenomen via Google afbeeldingen van www.skep.be - Dysplasieën of andere afwijkingen in de structuur van de hersenen zijn bij MRI onderzoek niet aangetoond.
- Aanwijzingen voor een ontsteking van hersenweefsel zijn niet gevonden, met name niet in het hersenweefsel dat bij operatie is uitgenomen.
- Een auto-immuun aandoening is overwogen. Inderdaad zijn bij sommige patiënten antilichamen tegen hersenweefsel aangetoond maar deze komen ook voor bij andere patiënten en bij patiënten zonder LKS (Nieuwenhuis & Nicolai, 2006).
- De eeg-afwijkingen bij LKS zijn verwant aan die van een ander syndroom dat ook op kinderleeftijd ontstaat en ook tot neuropsychologische stoornissen leidt. Dit andere syndroom draagt de naam van de eeg-afwijkingen die het kenmerken: “continue pieken en golven tijdens non-REM slaap” (Engels: continuous spikes and waves during sleep: CSWS). Sommige kinderen met een atypische vorm van Rolandische epilepsie (zie Ziektebeschrijving Rolandische epilepsie) krijgen LKS. Ook is verwantschap van LKS met autisme geopperd.
- Van een genetisch identieke eeneiige tweeling had het ene kind LKS en het andere niet. Dit verschil sluit genetische predispositie niet uit, maar suggereert dat nog een andere factor van belang is (Nieuwenhuis & Nicolai, 2006).
Ziekteverschijnselen
ZiekteverschijnselenVerschijnselen
1. Ouders merken dat hun overigens gezonde kind geleidelijk steeds minder op spreken reageert: het lijkt doof te worden. Het verschijnsel kan ook abrupt ontstaan (Duran et al., 2009). Bij onderzoek blijkt het gehoor intact; het kind begrijpt gesproken taal niet meer (verbale agnosie). Soms worden ook andere geluiden niet meer begrepen (auditieve agnosie). Gelijktijdig of na enige tijd is het kind niet meer in staat woorden of in ernstige gevallen zelfs geluiden te uiten: het kind is mutistisch geworden. Men veronderstelt dat het mutisme secundair is aan het verlies van begrip voor gesproken woorden.
2. Zeventig tot tachtig procent van de kinderen heeft één of meer epileptische aanvallen. Deze kunnen zich eerder, voor, of ook na ontstaan van de taalstoornis voordoen. De aanvallen kunnen partieel (trekkingen in een deel van het lichaam) zijn en alleen ’s nachts voorkomen, of ze zijn gegeneraliseerd: trekkingen in het gehele lichaam, of absences.
3. Onderzoek van de elektrische potentiaalveranderingen aan het oppervlak van de hersenen (het eeg) toont bij het slapende kind ontladingen met pieken en langzame golven boven de temporale kwabben; de ontladingen hebben hun origine eenzijdig in het bovenste deel van de temporale schors, in het gebied dat betrokken is bij de integratie van gehoors- en andere zintuiglijke informatie (Metz-Lutz, 2009; Castillo et al., 2008).
4. Het gedrag van het kind verandert. Hyperactiviteit, verlies van initiatief, agressiviteit, woedeaanvallen, slaapstoornissen en zelfs psychotische episoden kunnen zich voordoen. Ten dele is het veranderde gedrag reactief, een reactie op de ellende die het doormaakt; een relatie met eeg-afwijkingen in het voorste, frontale hersengebied is ook gesuggereerd (Robinson et al., 2001).
Beloop
Op de leeftijd van drie jaar zijn de meeste normaal ontwikkelende kinderen vertrouwd met de karakteristieke klanken van de taal die in hun omgeving wordt gesproken. De ontwikkeling van taal en spraak is bij sommige kinderen voorafgaande aan duidelijke verschijnselen van LKS traag verlopen (zie bijvoorbeeld Sinclair en Snyder, 2005). Ergens tussen het derde en zevende jaar worden meestal de eerste verschijnselen opgemerkt. Daarna wisselen perioden van verbetering en verergering elkaar af (Nieuwenhuis en Nicolai, 2006). Gemiddeld duurt het ongeveer vier jaren voordat enig blijvend herstel intreedt, veelal na het tiende jaar. De eeg-afwijkingen verminderen of verdwijnen, de communicatie verbetert, de kinderen gaan meer geluiden begrijpen, het gedrag verbetert. Adolescenten en jongvolwassenen die alleen behandeld zijn met anti-epileptica zijn niet altijd vrij van epileptische aanvallen en houden vaak ernstige taalstoornissen (Duran et al., 2009; Robinson et al., 2001).
Cognitief functioneren
Cognitief functionerenIntelligentie
Het verbale intelligentiequotiënt (VIQ)is gestoord als gevolg van de verbale agnosie. In de regel is het performale intelligentiequotiënt (PIQ) bij de kleine aantallen kinderen die daarop zijn onderzocht binnen de grenzen van het normale (Robinson et al., 2001).
Andere cognitieve functies
LKS belemmert het leerproces ernstig. Het lezen blijft langzaam en inefficiënt ook van kinderen die voorafgaande aan het ontstaan van LKS hebben leren lezen. Goed herstelde adolescenten met een normale woordenschat, houden toch moeite met nazeggen van zinnen, woorden en zinloze lettergrepen (Metz-Lutz, 2009; Sieratzki et al., 2001).
Medische behandeling
Medische behandeling• De epileptische aanvallen verdwijnen snel na starten van behandeling met anti-epileptica, de taalfunctiestoornissen niet.
• Bij sommige kinderen met LKS worden om verbreiding van de epileptische ontladingen in de schors tegen te gaan sneetjes in de hersenschors aangebracht die de verbindingen in het horizontale vlak tussen nabijgelegen gebieden onderbreken met behoud van de verbindingen met dieper liggend hersenweefsel (multipele subpiale transsecties, MST) (Castillo et al., 2008).
• Sommige kinderen met LKS worden behandeld met corticosteroïden, in de veronderstelling dat abnormale afweerreacties tegen het eigen lichaam een rol spelen (Sinclair & Snyder, 2005) .
• Van beide laatstgenoemde behandelingen zijn successen gemeld, maar de bewijskracht is tot nu toe zwak. De resultaten van behandeling met immuunglobulinen tenslotte zijn nog niet overtuigend (Arts et al., 2009).
Begeleiding
Begeleiding1. Neuropsychologisch onderzoek van de resterende taalfunctie en van andere cognitieve functies is nodig om tot een beleid te komen waarmee de stoornissen op de gebieden van taalfunctie, gnosis en leren gecompenseerd en gedragsproblemen voorkomen of tegengegaan worden.
2. Voor alles moet de communicatie met het kind gaande blijven of hersteld worden. Lukt dat niet via de spraak (kinderen met LKS tonen vaak aversie tegen stemmen en spraak), dan moeten non-verbale wegen bewandeld worden. Kinderen die al kunnen lezen hebben soms steun van geschreven woorden. Anders moet gecommuniceerd worden via bijvoorbeeld plaatjes of pictogrammen. Lukt dat niet, dan stapt men het beste zo snel mogelijk over op aanleren van gebarentaal zoals doven gebruiken of zoals voor kinderen met bijvoorbeeld autisme wordt aangewend. Kinderen met LKS hebben met het leren van gebarentaal over het algemeen niet veel moeite en kennis van gebarentaal staat latere terugkeer naar mondeling taalgebruik niet in de weg. Er zijn wel enkele voorwaarden: het kind moet niet dyspractisch zijn, de ouders moeten het met de overgang op gebarentaal eens zijn en (één van hen moet) de gelegenheid hebben deze zelf ook te leren en er moet een communicatietherapeut zijn die het kind en belangrijke personen in zijn omgeving de gebarentaal kan leren. Bij herstel van communicatie verbetert meestal ook het gedrag van het kind (Deonna et al., 2009) .
Een kind met LKS
Een kind met LKS fajenn 29 mei, 2010 - 13:19Literatuur
LiteratuurArts WFM , Aarsen FK, Scheltens-de Boer M, Catsman-Berrevoets CE (2009) Landau-Kleffner syndrome and CSWS syndrome: treatment with intravenous immunoglobulins. Epilepsia 50, supplement 7: 55-58
Castillo EM, Butler IJ, Baumgartner JE, Passaro A, Papanicolaou AC (2008) When epilepsy interferes with word comprehension: Findings in Landau Kleffner syndrome. Journal of Child Neurology 23: 97-101
Deonna T, Prelaz-Girod A-C, Mayor-Dubois C, Roulet-Perez E (2009) Sign language in Landau-Kleffner syndrome. Epilepsia 50, Suppl 7, 77-82
Duran MHC, Guimaräes A, Medeiros LL, Guerreiro MM (2009) Landau-Kleffner syndrome: long-term follow-up Brain & Development 31: 58-63
Jennekens-Schinkel A & Jennekens FGI (2008) Neuropsychologie van neurologische aandoeningen in de kindertijd. Amsterdam: Uitgeverij Boom, pp 426-427
Metz-Lutz MN (2009) The assessment of auditory function in CSWS: Lessons from long-term outcome. Epilepsia 50, supplement 7: 73-77
Nieuwenhuis L, Nicolai J (2006) The pathophysiological mechanisms of cognitive and behavioral disturbances in children with Landau-Kleffner syndrome or epilepsy with continuous spike-and-waves during slow-wave sleep. Seizure 15: 249-258
Robinson RO, Baird G, Robinson G, Simonoff E (2001) Landau-Kleffner syndrome: course and correlates with outcome. Developmental Medicine & Child Neurology 43: 243-247
Sinclair DB, Snyder TJ (2005) Corticosteroids for the treatment of Landau-Kleffner syndrome and continuous spike-wave discharge during sleep. Pediatric Neurology 32: 300-306
Sieratzki JS, Calvert GA, Brammer M, David B, Woll LC (2001) Accessibility of spoken, written and sign language in Landau-Kleffner syndrome: a linguistic and functional MRI study. Epileptic Disorders 3: 79-89
Kwaadaardige epilepsieën
Kwaadaardige epilepsieën fajenn 29 mei, 2010 - 13:26Inleiding
InleidingHersenaandoeningen kunnen al bij zeer jonge kinderen leiden tot ernstige vormen van epilepsie die met stoornissen van cognitie en gedrag gepaard gaan.Het ontwikkelingsstadium van de grote hersenen bepaalt voor een deel de ziekteverschijnselen. In de beginfasen is het onderscheid met zeldzame, goedaardiger beelden vaak niet eenvoudig. Over het algemeen is bij deze kwaadaardige ziekten meer bekend over het acute probleem van de epileptische aanvallen dan over de stoornissen die zich – vaak na verloop van tijd – manifesteren in cognitie en gedrag.
Andere teksten op internet
Andere teksten op internetwww.kinderneurologie.eu biedt gedetailleerde en goed toegankelijke Nederlandstalige informatie over een groot aantal epilepsiesyndromen, waaronder kwaadaardige.
www.ilae-epilepsy.org/ctf/ is de Engelstalige website van het internationale verbond tegen epilepsie met medische informatie over een reeks van epilepsiesyndromen.
Voor Nederlandstalige websites met praktische voorlichting aan ouders en met informatie over contact tussen ouders, zie de Ziektebeschrijving Epilepsie, andere teksten op internet en Ziektebeschrijving Epilepsie, Begeleiding.
Beginnend in eerste drie levensmaanden, myoklonische encefalopathie, syndroom van Ohtahara
Beginnend in eerste drie levensmaanden, myoklonische encefalopathie, syndroom van OhtaharaMyoklonische encefalopathie [early myoclonic epileptic encephalopathy (EMEE)] en het syndroom van Ohtahara
Epidemiologie en oorzaak
Beide aandoeningen zijn zeldzaam (Zupanc 2009; Ohtahara & Yamatogi, 2006). In Nederland is voor zover wij weten over geen van beide gerapporteerd. Hoe vaak de syndromen in Nederland voorkomen is niet bekend. Ze ontstaan in de eerste levensmaanden bij kinderen met ernstige aangeboren hersenafwijkingen, stofwisselingsstoornissen (bij Ohtahara zeer zelden) of op basis van een genetische afwijking. Een behandelbare mitochondriale aandoening is bij uitzondering ontdekt bij een kind met het syndroom van Ohtahara. EMEE is vaak familiair.
Verschijnselen
Kinderen met het syndroom van Ohtahara hebben dagelijks 100 tot 300 spasmen waarbij het gehele lichaam of een deel van het lichaam betrokken is. Kinderen met EMEE hebben vooral veel kleine spierschokjes; grotere trekkingen komen ook voor.
Het lukt vaak niet de aanvallen met anti-epileptica te onderdrukken.
De ontwikkeling van de kinderen is sterk vertraagd. Het syndroom van Ohtahara kan overgaan in het syndroom van West (zie elders in deze tekst). De kinderen overlijden of ze blijven mentaal ernstig gehandicapt en worden meestal te eniger tijd opgenomen in een verpleeghuis.
Cognitie en gedrag
Sommige kinderen komen in aanmerking voor evaluatie van hun cognitieve mogelijkheden of voor advies betreffende gedragsproblemen.
Beginnend in eerste levensjaar. Syndromen van Dravet en West
Beginnend in eerste levensjaar. Syndromen van Dravet en WestErnstige myoklonische epilepsie van jonge kinderen [severe myoclonic epilepsy of infancy, SMEI)] of syndroom van Dravet
Epidemiologie en oorzaak
SMEI ontstaat bij naar schatting twee tot vier per 100.000 kinderen per jaar. De aandoening is bij de meeste kinderen (omstreeks 80%) te wijten aan een afwijking in een ionkanaal in zenuwcelmembranen waar natriumionen door kunnen passeren (Stafstrom, 2009; Wheless, 2009). De afwijking is het gevolg van een mutatie in het gen dat de code bevat voor het eiwit dat het ionkanaal vormt. Afhankelijk van de aard van de mutatie verschillen de gevolgen voor het ionkanaal, en daarmee variëren de ziekteverschijnselen. MRI toont in de beginfase geen structuurafwijkingen van de hersenen. Koortsstuipen komen bij een grote minderheid van de kinderen familiair voor.
Epileptische aanvallen
Kenmerkend voor het syndroom is dat het kind aanvankelijk een ongestoorde ontwikkeling doormaakt. De eerste aanval doet zich voor tussen de derde en elfde maand, vooral omstreeks de vijfde tot zevende maand. Veelal hebben de kinderen dan koorts. De aanval kan langdurig zijn en zelfs langer duren dan 30 minuten (status epilepticus zie deze website, "Epilepsie", de paragraaf over "Aanvallen").De trekkingen kunnen gegeneraliseerd voorkomen of in een lichaamshelft ("hemiconvulsies"). In de periode daarna volgen verschillende typen epileptische aanvallen, ook als het kind geen koorts heeft: gegeneraliseerd klonisch of tonisch en klonisch, klonisch in een lichaamshelft of in een deel van een lichaamshelft, myoklonieën, en wegrakingen (absences). Niet zelden volgt de ene aanval de andere op (status-epilepticus) (Ragona et al., 2010). De moeilijk behandelbare aanvallen kunnen worden uitgelokt door verhoging van lichaamstemperatuur (warm badwater), licht, staren naar bijvoorbeeld drukke patronen of anderszins. Myoclonieën en absences verminderen of verdwijnen in de loop van jaren bij veel adolescenten en jonge volwassenen. De aanvallen doen zich in meerderheid vooral ’s nachts voor. Een minderheid van de kinderen wordt op volwassen leeftijd aanvalsvrij.
Cognitie en gedrag
Vertraging, stilstand of geleidelijke achteruitgang van de psychomotore ontwikkeling manifesteert zich omstreeks het tweede jaar of daarna (Akiyama et al., 2010; Ragona et al., 2010). Onderzoek heeft aannemelijk gemaakt dat dit afwijkende ontwikkelingsbeloop en de epilepsie het gevolg zijn van de genetische afwijking (Riva et al., 2009). Bij in totaal 14 kinderen tussen 4 en 13 jaar varieerde het ontwikkelingsquotiënt van 12 kinderen tussen 20 en 40, bij twee anderen tussen 60 en 80 (Wolff et al., 2007). De "mentale retardatie" kan, zoals aangetoond bij een groep van 16 kinderen van 10 jaar en ouder licht tot matig zijn maar is vaker ernstig (Ragona et al., 2010). Op volwassen leeftijd is een minderheid niet in staat tot het gebruik van taal (Akiyama et al., 2010).
Bij veel kinderen verandert het gedrag: er ontstaan hyperactiviteit, aandachtstoornissen, gestoord contact en soms oppositioneel gedrag.
Andere stoornissen
Gedurende het beloop van de ziekte blijken nog andere neurologische afwijkingen, vooral van de motoriek (ataxie, tekenen van een piramidebaansyndroom, tremoren) (Ragona et al., 2010; Nolan et al., 2008). Er is risico op vervroegd overlijden, tijdens status epilepticus, door longontsteking of door onduidelijke oorzaak (Akiyama et al., 2010; Wolff et al., 2006).
Medische behandeling en begeleiding
De kinderen worden behandeld met anti-epileptica of met een ketogeen dieet (zie Ziektebeschrijving Epilepsie, behandeling . Aanvalsvrijheid wordt bij de meeste kinderen niet bereikt. Het anti-epilepticum stiripentol lijkt het meest effectief te zijn (Chiron et al., 2000; Inoue et al., 2009). Deze conclusie steunt mede op een gecontroleerd onderzoek van de effectiviteit van valproïnezuur en clobazam in combinatie met stiripentol (Chiron et al., 2000). Of stiripentol behalve de epileptische aanvallen ook de cognitieve achteruitgang afremt is niet bekend. Van sommige andere anti-epileptica wordt gesteld dat ze de aanvallen kunnen verergeren (carbamazepine, lamotrigine, vigabatrine en fenytoine) (Wheless, 2009).
Verzorgers van kinderen met SMEI dienen voorbereid te zijn op mogelijk ontstaan van status epilepticus. Een protocol voor noodgevallen kan hen daarbij helpen. Een ouder die thuis de zorg heeft voor een kind met dit syndroom zou, bij voorkeur dagelijks, enige tijd vrijaf moeten hebben om de verzorgingslast in de resterende tijd beter te kunnen dragen (Nolan et al., 2008).
Vaststelling van de sterke en zwakke kanten van het kind kan, zeker in de fase van stagnerend en achteruitgaand cognitief en gedragsmatig functioneren, ouders en/of verzorgers richtlijnen opleveren voor de begeleiding van het kind. Ook eventueel gunstige invloed van behandeling met bijvoorbeeld anti-epileptica op de cognitieve ontwikkeling dient aan de hand van onderzoek te worden vastgesteld.
Syndroom van West (“Infantile spasms”)
Epidemiologie en oorzaak
Het syndroom van West ontstaat bij ongeveer 3 per 10.000 geborenen, vooral in de periode van de vierde tot de achtste levensmaand (Zupanc, 2009; Guerrini, 2006), en iets meer bij jongens dan bij meisjes. Het syndroom kan zich voordoen bij een groot aantal ernstige hersenafwijkingen. Deze variëren van aangeboren misvormingen van en in de hersenen, hersenbeschadiging door zuurstofnood bij de geboorte, tot stofwisselingsstoornissen in de hersenen. Bij een minderheid van de kinderen kunnen structuurafwijkingen van de hersenen niet worden vastgesteld; een aantal van hen heeft een achterhaalbare genetische afwijking (Marshall et al., 2008).
Verschijnselen
De ontwikkeling blijft bij de meerderheid van de kinderen al achter voor er aanvallen zijn. Spasmen treden zeer plotseling op en duren kort. Vaak herhalen ze zich een aantal keren, zodat ze groepsgewijze voorkomen. Het kind buigt de hals of strekt de nek met gelijktijdig heffen en spreiden van de armpjes, of juist tegen het lichaam drukken van de armpjes. Een spasme kan weinig meer zijn dan een knik van het hoofd maar het kan ook een samentrekking van vrijwel het gehele lichaam zijn. Asymmetrie wijst op een eenzijdige hersenafwijking. De spasmen worden niet altijd snel herkend als afwijkend. De sociale reacties verminderen. Het kind lacht niet meer en reageert niet meer op visuele of geluidsprikkels. Het elektro-encefalogram (eeg) toont een zeer chaotisch beeld (hypsaritmie); alleen bij heel ernstige hersenstoornissen kan het eeg afwijken van dit kenmerkende beeld.
Medische behandeling
Over de beste behandeling bestaat onzekerheid. Adrenocorticotroop hormoon (ACTH) en vigabatrine komen meer in aanmerking dan de gebruikelijke anti-epileptica. Verdwijnen van de spasmen en afwezigheid van afwijkingen op de MRI van de hersenen zijn prognostisch gunstig. Operatieve behandeling wordt overwogen als de hersenafwijking eenzijdig is. De grote meerderheid van de kinderen houdt epileptische aanvallen en blijft cognitief gehandicapt (Partikian & Mitchell, 2009; Jennekens-Schinkel & Jennekens, 2008).
Cognitie en gedrag
De meeste kinderen met het syndroom van West hebben een verstandelijke beperking die kan variëren van diep tot licht. Wanneer beeldvormend onderzoek geen hersenafwijkingen toont is de kans groter dat het kind cognitief “normaal” functioneert, dat wil zeggen dat het een IQ heeft boven 74.
Neuropsychologische diagnostiek kan houvast bieden voor begeleiding. De vooruitzichten hangen vooral af van de ernst van de ziekte en het al dan niet progressieve karakter van de hersenaandoening.
Beginnend na eerste levensjaar. Syndromen van Lennox Gastaut en Doose
Beginnend na eerste levensjaar. Syndromen van Lennox Gastaut en DooseSyndromen van Lennox Gastaut (LGS) en van Doose
Epidemiologie en oorzaak
Over incidentie en prevalentie van LGS bestaat onzekerheid; van alle vormen van epilepsie bij kinderen zou LGS drie tot vijf procent uitmaken (Guerrini, 2006; Trevathan, 2002). De kinderen zijn bij de eerste verschijnselen in de regel jonger dan acht jaar, meestal tussen drie en vijf jaar. Bij de meerderheid van de kinderen ontstaat het syndroom secundair aan een al langer aanwezige hersenaandoening, bijvoorbeeld een dysplasie of cerebrale parese (zie Ziektebeschrijving Cerebrale parese).
Verschijnselen
LGS is gekarakteriseerd door een verscheidenheid van epileptische aanvallen, opvallende eeg-afwijkingen en cognitieve stoornissen. Het voor LGS meest kenmerkende type aanval is een tonische contractie die seconden tot een minuut kan aanhouden en waarbij hoofd en romp worden gebogen, de armen worden gespreid en geheven, en die soms wordt voorafgegaan door een kreet of gil. Andere aanvallen hebben het karakter van, soms in reeksen achter elkaar optredende, absences, van plotseling vallen door atonie of juist tonische contractie van spieren, en van myoklonieën. Al voor de LGS zich manifesteert zijn de meeste kinderen door de al bestaande hersenafwijkingen in ontwikkeling vertraagd. Het percentage cognitief gestoorde kinderen stijgt in de jaren na het begin van de aanvallen geleidelijk tot 75 en groter. Veel nadere gegevens zijn hierover niet beschikbaar. Kinderen zonder al voor de LGS bestaande hersenafwijkingen ontwikkelen tot de eerste verschijnselen van LGS normaal; hun ziektebeeld kan lijken op het syndroom van Doose (myoklonische astatische epilepsie).
Bij het syndroom van Doose komen ook verschillende type aanvallen voor, waaronder aanvallen die de kinderen doen vallen. Het beloop is in sommige gevallen gunstig en in andere ongunstig (Zupanc, 2009; Guerrini, 2006). In hoeverre LGS zonder hersenafwijkingen genetisch verschilt van het syndroom van Doose zal nog moeten blijken.
Medische behandeling
De epilepsie wordt behandeld met anti-epileptica (felbamaat, lamotrigine, topiramaat) of ook wel met corticosteroïden of adrenocorticotroop hormoon (ACTH). Tachtig procent van de kinderen houdt aanvallen. Het effect van de behandeling op het cognitieve functioneren is niet geëvalueerd (Arzimanoglou et al., 2009; Jennekens-Schinkel & Jennekens, 2008).
Een kind met het syndroom van West
Een kind met het syndroom van West fajenn 29 mei, 2010 - 13:36Literatuur
LiteratuurAkiyama M, Kobayashi K, Yoshinaga H, Ohtsuka Y (2010) A long-term follow-up study of Dravet syndrome up to adulthood. Epilepsia 51: 1043-1052
Arzimanoglou A, French J, Blume WT, Cross JH, Ernst J-P, Feucht M, Genton P, Guerrini R, Kluger G, Pellock JM, Perucca E, Wheless JW (2009) Lennox-Gastaut syndrome: a consensus approach on diagnosis, assessment, management, and trial methodology. Lancet Neurology 8: 82-93
Chiron C, Marchand MC, Tran A, Rey E, d'Athis P, Vincent J, Dulac O, Pons G (2000) Stiripentol in severe myoclonic epilepsy in infancy: a randomised placebo-controlled syndrome-dedicated trial. STICLO study group. Lancet 356: 1638-1642
Guerrini R (2006) Epilepsy in children. Lancet 367: 499-524
Inoue Y, Ohtsuka Y, Oguni H, Tohyama J, Baba H, Fukushima K, Ohtani H, Takahashi Y, Ikeda S (2009) Stiripentol open study in Japanese patients with Dravet syndrome. Epilepsia 50: 2362-2368
Jennekens-Schinkel A & Jennekens FGI (2008) Neuropsychologie van neurologische aandoeningen in de kindertijd. Amsterdam: Boom, p 425 en pp 429-430
Korff C, Laux L, Kelley K, Goldstein J, Koh S, Nordli Jr D (2007) Dravet syndrome (severe myoclonic epilepsy in infancy): a retrospective study of 16 patients. Journal of Child Neurology 22: 185-194
Marshall CR en 24 andere auteurs (2008) Infantile spasms is associated with deletion of the MAG12 gene on chromosome 7q11.23-q21.11. The American Journal of Human Genetics 83: 106-111
Nolan K, Camfield CS, Camfield PR (2008) Coping with a child with Dravet syndrome: insights from families. Journal of Child Neurology 23: 690-694
Ohtahara S, Yamatogi Y (2006) Ohtahara syndrome: with special reference to its developmental aspects for differentiating from early myoclonic encephalopathy. Epilepsy Research 70Suppl1: S58-S67
Partikian A, Mitchell WG (2009) Neurodevelopmental and epilepsy outcomes in a North American Cohort of patients with infantile spasms. Journal of Child Neurology, DOI 10.1177/0883073809341664
Ragona F, Brazzo D, De Giorgo I, Morbi M, Freri E, Teutonico F en 5 andere auteurs (2010) Dravet syndrome: Early clinical manifestations and cognitive outcome in 37 Italian patients. Brain & Development 32: 71-77
Riva D, Vago C, Pantaleoni C, Bulgheroni S, Mantegazza M, Franceschetti S (2009) Progressive neurocognitive decline in two children with Dravet syndrome, De Novo SCN1A truncations and different epileptic phenotypes. American Journal of Medical Genetics Part A 149A: 2339-2345
Stafstrom CE (2009) Severe epilepsy syndromes of early childhood: the link between genetics and pathophysiology with a focus on SCN1A mutations. Journal of Child Neurology 24, supplement 1: 15S-23S
Trevathan E (2002) Infantile spasms and Lennox-Gastaut Syndrome. Journal of Child Neurology 17, Supplement 2: S009-S022
Wheless JW (2009) Managing severe epilepsy syndromes of early childhood. Journal of Child Neurology 24, supplement 1: 24S-32S
Wolff M, Cassé-Perrot C, Dravet C (2006) Severe myoclonic epilepsy of infants (Dravet syndrome): natural history and neuropsychological findings. Epilepsia 47: 45-48
Zupanc M (2009) Clinical evaluation and diagnosis of severe epilepsy syndromes of early childhood. Journal of Child Neurology 24, Supplement 1: 6S-14S